Deep learning reveals predictive sequence concepts within immune repertoires to immunotherapy

Abstract
Background: Checkpoint inhibition (CPI) in cancer has changed the landscape of how oncologists treat advanced disease [1,2,3,4,5,6]; while there has been tremendous promise of immunotherapy, most patients do not respond to treatment [6]. Biomarker development has grown as the field attempts to better select patients that may benefit from immunotherapy as well as to further understanding of effective use of CPI in cancer [7,8,9,10]. One area of interest has been studying the T-cell response through TCR-sequencing, allowing for a characterization of the antigenic determinants of response [10,11,12]. However, much of the work has been limited to characterizing the quantitative aspects of the TCR repertoire. Here, for the first time, we queried whether there are TCR sequence concepts (i.e. motifs) that were predictive of response to immunotherapy. Methods: We employ DeepTCR [13], a previously described set of deep learning algorithms, to search for sequence concepts in pre-treatment tumor samples that are predictive of effective immunotherapy in CheckMate 038 (NCT01621490), a clinical trial of CPI. We fit DeepTCR’s multiple instance TCR repertoire classifier (Fig. 1) to predict response (via RECIST) in this cohort and assess not only the predictive performance of the model but insights into an effective antigen-specific T-cell response during CPI. Results: When applying DeepTCR to predict response, a joint representation of TCR repertoire with HLA background of the patient outperformed models that used TCR sequence or HLA genotype information alone (Fig. 2a). This model’s predictions of likelihood to respond to treatment also significantly stratified progression free survival in this cohort of patients (Fig. 2b). For more qualitative descriptions of the TCR repertoire that defined an effective immune response, we used DeepTCR’s variational autoencoder (VAE) to construct an unsupervised representation of the TCR sequences and highlighted the most predictive sequences for responders (blue) and non-responders (red) (Fig. 3 a,b). We noted that not only are the distributions within responders and non-responders multi-modal, but these multiple modes are shared between patients. When comparing the predictive signature in pre- vs post-treatment repertoires, we noted that while the responder signature remained constant over the course of treatment, the non-responder signature demonstrated changes in the TCR sequence space (Fig. 3 c,d). Conclusions: Taken together, these findings highlight the utility of deep learning to identify sequence features of TCR repertoire under the influence of immunotherapy and note that the pre-existing antigenic response is a key predictor of response to treatment and the maintenance of this antigenic response is a hallmark of clinical benefit. Ethics Approval: CheckMate 038 (NCT01621490) is a BMS-sponsored, multi-center, institutional-review-board-approved, phase 1 biomarker study of nivolumab, ipilimumab, and nivolumab in combination with ipilimumab in patients with advanced melanoma.
Presentation Video
Figure 1
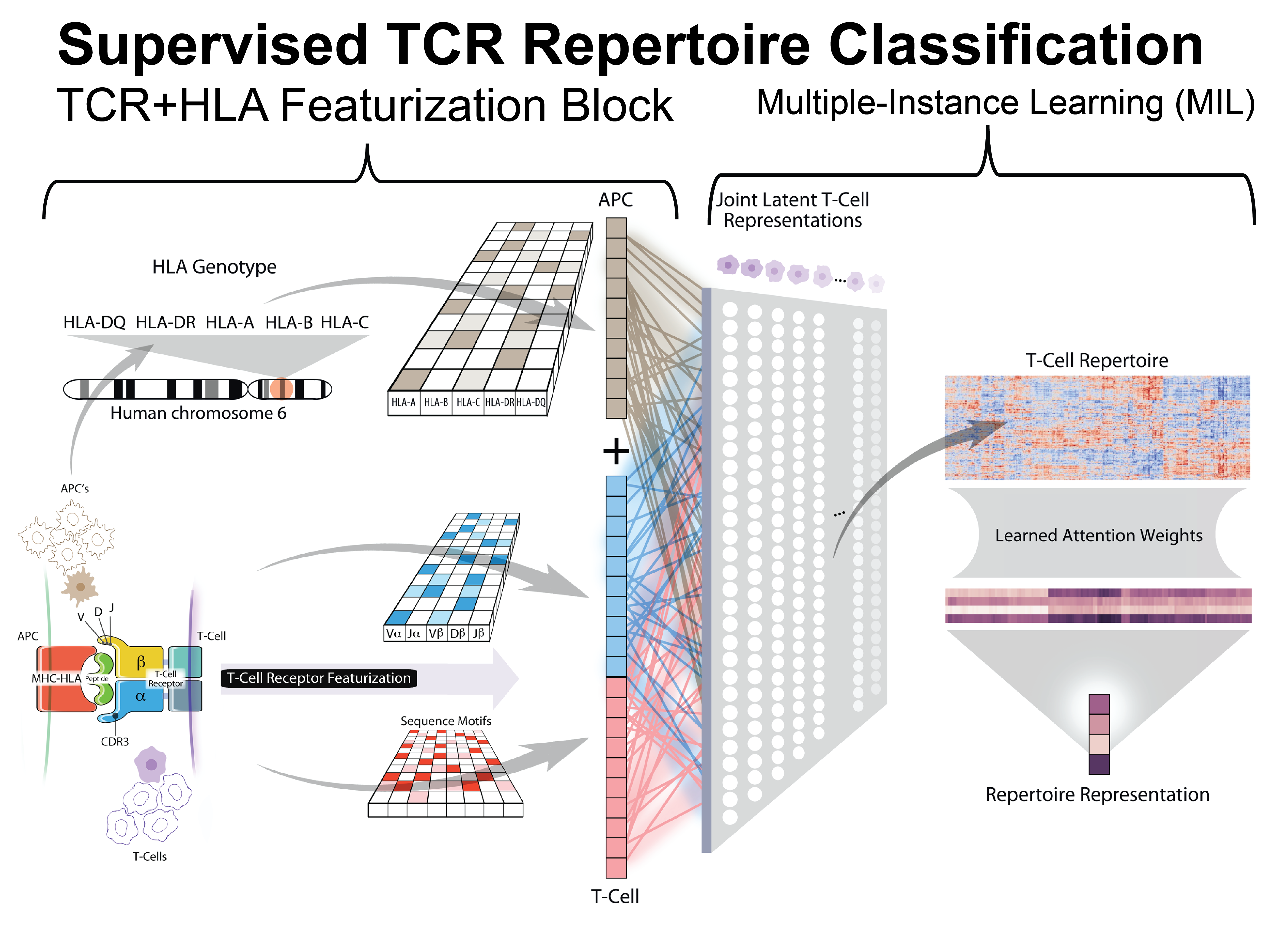 DeepTCR’s Multiple Instance Learning Repertoire Classifier
We expand on previous work by modifying the DeepTCR Featurization block to incorporate the HLA background within which a given collection of TCRs were observed within. The HLA background of a sample/individual is provided to the neural network in a multi-hot representation that is rerepresented in a learned continuous embedding layer and concatenated to the continuous learned representation of the TCR. As previously described, we implement a multi-head attention mechanism to make sequence assignments to concepts within the sample. The number of concepts in the model is a hyperparameter, which can be varied by the user depending on the heterogeneity expected in the repertoires. Of note, this assignment of a sequence to a concept is done through an adaptive activation function that outputs a value between 0 and 1, allowing the network to put attention on the sequences that are relevant to the learning task. When taking the average of these assignments over all the cells in a repertoire, this results in a value within the neural network that directly corresponds to the proportion of the repertoire that is described by that learned concept. These proportions of concepts in the repertoire are then sent into a final traditional classification layer.
DeepTCR’s Multiple Instance Learning Repertoire Classifier
We expand on previous work by modifying the DeepTCR Featurization block to incorporate the HLA background within which a given collection of TCRs were observed within. The HLA background of a sample/individual is provided to the neural network in a multi-hot representation that is rerepresented in a learned continuous embedding layer and concatenated to the continuous learned representation of the TCR. As previously described, we implement a multi-head attention mechanism to make sequence assignments to concepts within the sample. The number of concepts in the model is a hyperparameter, which can be varied by the user depending on the heterogeneity expected in the repertoires. Of note, this assignment of a sequence to a concept is done through an adaptive activation function that outputs a value between 0 and 1, allowing the network to put attention on the sequences that are relevant to the learning task. When taking the average of these assignments over all the cells in a repertoire, this results in a value within the neural network that directly corresponds to the proportion of the repertoire that is described by that learned concept. These proportions of concepts in the repertoire are then sent into a final traditional classification layer.
Figure 2
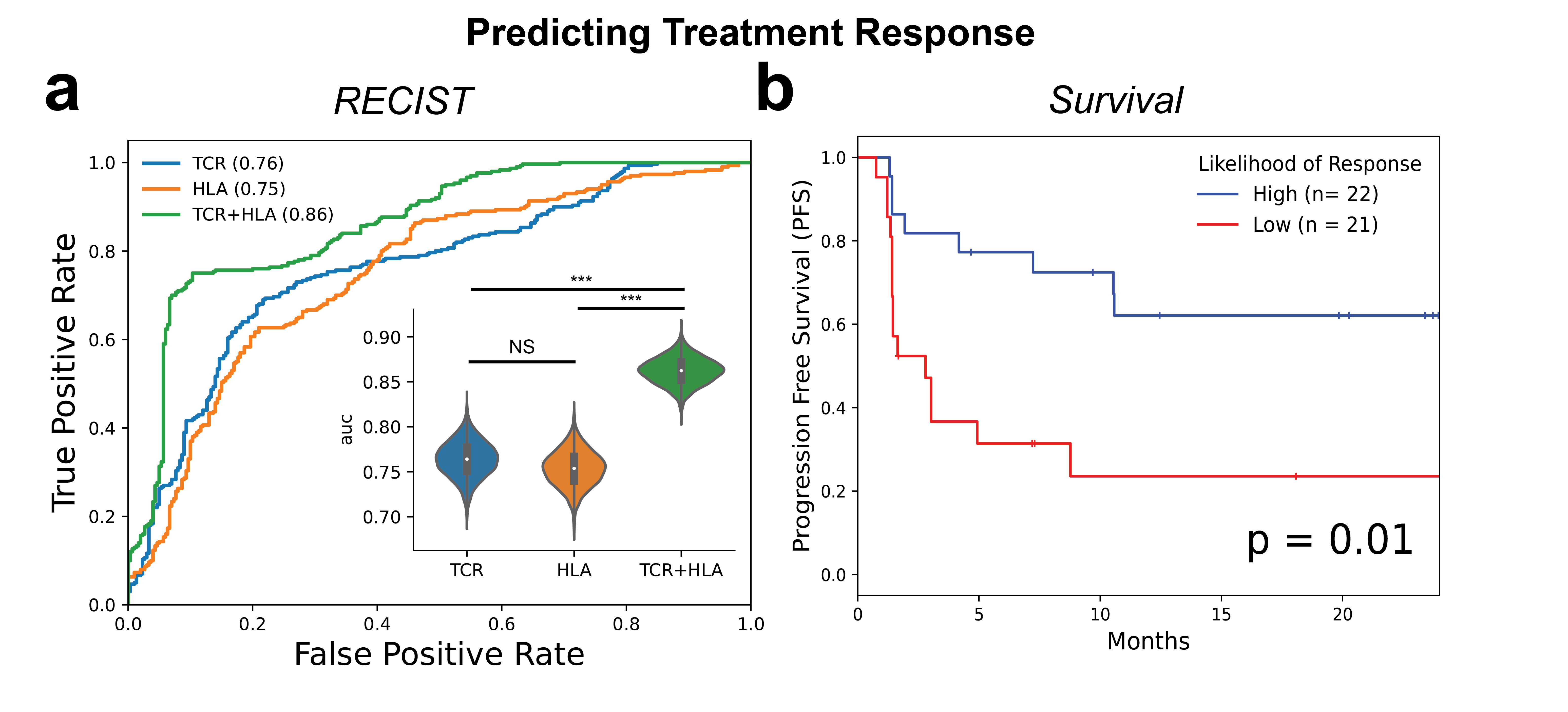 Repertoire Classification in Pre-Treatment TIL
a) Pre-treatment tumor biopsies were collected and TCR-Seq was performed from 43 patients enrolled in the CheckMate-038 (parts 2-4) clinical trial where they were either treated with anti-PD1 monotherapy (9 patients) or anti-PD1+ anti-CTLA combination therapy (34 patients) and followed for radiographic response to therapy via RECIST v1.1. Complete Responders and Partial Responders (CRPR) were denoted as responders to therapy while Stable Disease and Progressive Disease (SDPD) were denoted as nonresponders to therapy. Receiver Operating Characteristics (ROC) Curves were created for predicting response (complete response, partial response) to immunotherapy given either TCR, HLA, or TCR+HLA information to the supervised repertoire classifier (100 Monte-Carlo simulations with train size: 37, test size: 6). Bootstrap analyses (5000 iterations) were performed to construct confidence intervals (CI) around AUC values and assess differences in model performance, in which each AUC per sampling was compared in a paired manner across the three models designed above. The null hypothesis of two models exhibiting equivalent performance was rejected if the bootstrap difference did not cross 0. (*** : 99.9% CI). b) The likelihood of response generated by the TCR+HLA model was dichotomized into “High” and “Low” using the median predicted value in this cohort (taken over the MC test sets and averaged per sample) and the Kaplan-Meier (KM) curves were shown for progression free survival (PFS), log-rank p-value = 0.005.
Repertoire Classification in Pre-Treatment TIL
a) Pre-treatment tumor biopsies were collected and TCR-Seq was performed from 43 patients enrolled in the CheckMate-038 (parts 2-4) clinical trial where they were either treated with anti-PD1 monotherapy (9 patients) or anti-PD1+ anti-CTLA combination therapy (34 patients) and followed for radiographic response to therapy via RECIST v1.1. Complete Responders and Partial Responders (CRPR) were denoted as responders to therapy while Stable Disease and Progressive Disease (SDPD) were denoted as nonresponders to therapy. Receiver Operating Characteristics (ROC) Curves were created for predicting response (complete response, partial response) to immunotherapy given either TCR, HLA, or TCR+HLA information to the supervised repertoire classifier (100 Monte-Carlo simulations with train size: 37, test size: 6). Bootstrap analyses (5000 iterations) were performed to construct confidence intervals (CI) around AUC values and assess differences in model performance, in which each AUC per sampling was compared in a paired manner across the three models designed above. The null hypothesis of two models exhibiting equivalent performance was rejected if the bootstrap difference did not cross 0. (*** : 99.9% CI). b) The likelihood of response generated by the TCR+HLA model was dichotomized into “High” and “Low” using the median predicted value in this cohort (taken over the MC test sets and averaged per sample) and the Kaplan-Meier (KM) curves were shown for progression free survival (PFS), log-rank p-value = 0.005.
Figure 3
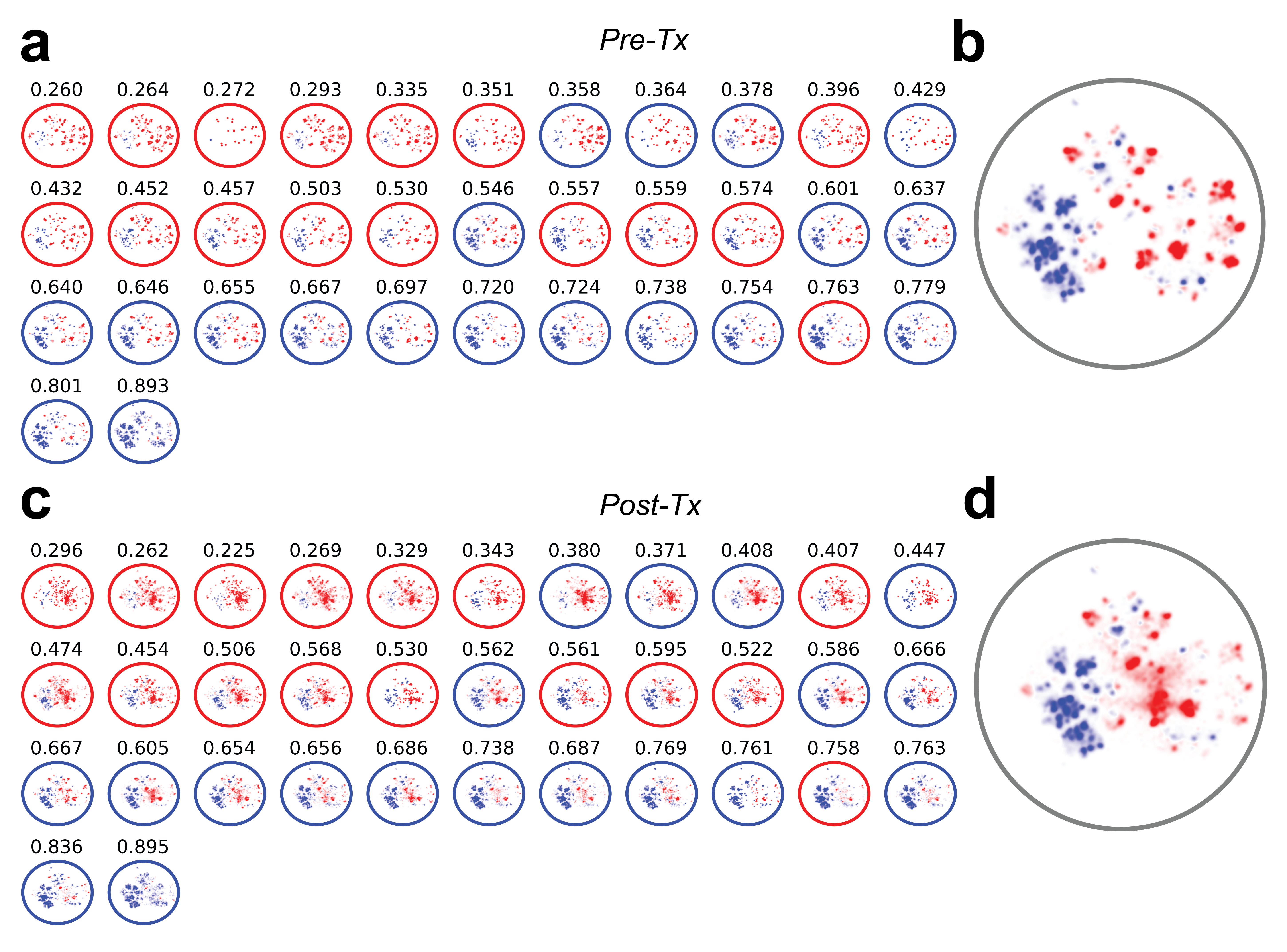 Pre vs Post-Treatment TCR Repertoire
In order to provide a descriptive understanding of the T-cell response in responders and non-responders in the CheckMate-038 clinical trial, we sought to characterize the distribution of the TCR repertoire in this cohort of patients. Data from CheckMate-038 were used to train a VAE on all sequence data (incorporating TCR+HLA information) in a sample and class agnostic fashion. The distribution of responders and non-responders repertoires were visualized via UMAP of the unsupervised VAE featurization. In order to visualize the distribution of the highly predictive TCR sequences, a per sequence prediction value was assessed following each MC simulation on the TCR’s within the independent test set, assigning the probability that a given TCR had a responder signature. Over the 100 MC simulations, each sequence in the cohort is assigned multiple prediction values that are averaged over all simulations to serve as a consensus predicted value for each sequence in this cohort of patients. Top 10% of sequences in responders and non-responders were selected and visualized over the entire cohort and on a per-sample basis where edge color denotes the ground truth label of the sample (non-responder = red, responder = blue) and average predicted likelihood taken over MC simulations to respond to treatment shown above each patient’s distribution. For each pair of pre/post treatment repertoires, the repertoire-level prediction was compared for pre vs. post treatment across all trained models and the top 10% of predictive sequences in the 35 paired pre/post repertoires were visualized across all paired samples (a & c) as well as over the entire cohort (b & d). (blue = most predictive of response, red = least predictive of response)
Pre vs Post-Treatment TCR Repertoire
In order to provide a descriptive understanding of the T-cell response in responders and non-responders in the CheckMate-038 clinical trial, we sought to characterize the distribution of the TCR repertoire in this cohort of patients. Data from CheckMate-038 were used to train a VAE on all sequence data (incorporating TCR+HLA information) in a sample and class agnostic fashion. The distribution of responders and non-responders repertoires were visualized via UMAP of the unsupervised VAE featurization. In order to visualize the distribution of the highly predictive TCR sequences, a per sequence prediction value was assessed following each MC simulation on the TCR’s within the independent test set, assigning the probability that a given TCR had a responder signature. Over the 100 MC simulations, each sequence in the cohort is assigned multiple prediction values that are averaged over all simulations to serve as a consensus predicted value for each sequence in this cohort of patients. Top 10% of sequences in responders and non-responders were selected and visualized over the entire cohort and on a per-sample basis where edge color denotes the ground truth label of the sample (non-responder = red, responder = blue) and average predicted likelihood taken over MC simulations to respond to treatment shown above each patient’s distribution. For each pair of pre/post treatment repertoires, the repertoire-level prediction was compared for pre vs. post treatment across all trained models and the top 10% of predictive sequences in the 35 paired pre/post repertoires were visualized across all paired samples (a & c) as well as over the entire cohort (b & d). (blue = most predictive of response, red = least predictive of response)