Probabilistic mixture models improve calibration of panel-derived tumor mutational burden in the context of both tumor-normal and tumor-only sequencing
Abstract
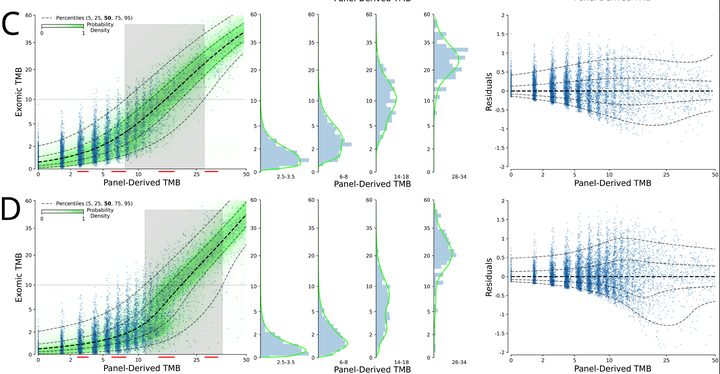
Background: Tumor mutational burden (TMB) has been investigated as a biomarker for immune checkpoint blockade (ICB) therapy. Increasingly, TMB is being estimated with gene panel-based assays (as opposed to full exome sequencing) and different gene panels cover overlapping but distinct genomic coordinates, making comparisons across panels difficult. Previous studies have suggested that standardization and calibration to exome-derived TMB be done for each panel to ensure comparability. With TMB cutoffs being developed from panel-based assays, there is a need to understand how to properly estimate exomic TMB values from different panel-based assays. Design: Our approach to calibration of panel-derived TMB to exomic TMB proposes the use of probabilistic mixture models that allow for nonlinear relationships along with heteroscedastic error. We examined various inputs including nonsynonymous, synonymous, and hotspot counts along with genetic ancestry. Using the TCGA cohort we generated a tumor-only version of the panel-restricted data by reintroducing private germline variants. Results: We were able to model more accurately the distribution of both tumor-normal and tumor-only data using the proposed probabilistic mixture models as compared to linear regression. Applying a model trained on tumor-normal data to tumor-only input results in biased TMB predictions. Including synonymous mutations resulted in better regression metrics across both data types, but ultimately a model able to dynamically weight the various input mutation types exhibited optimal performance. Including genetic ancestry improved model performance only in the context of tumor-only data, wherein private germline variants are observed.