Aggregation Tool for Genomic Concepts (ATGC): A deep learning framework for sparse genomic measures and its application to tumor mutational burden
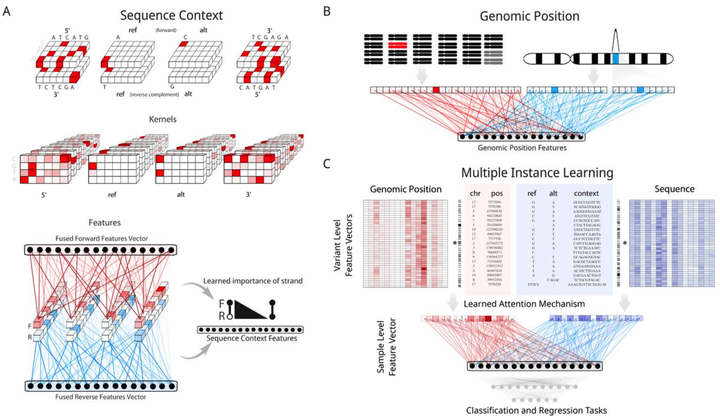
Abstract
Deep learning has the ability to extract meaningful features from data given enough training examples. Large scale genomic data are well suited for this class of machine learning algorithms; however, for many of these data the labels are at the level of the sample instead of at the level of the individual genomic measures. To leverage the power of deep learning for these types of data we turn to a multiple instance learning framework, and present an easily extensible tool built with TensorFlow and Keras. We show how this tool can be applied to somatic variants (featurizing genomic position and sequence context), and accurately classify samples according to whether they contain a specific variant (hotspot or tumor suppressor) or whether they contain a type of variant (microsatellite instability). We then apply our model to the calibration of tumor mutational burden (TMB), an increasingly important metric in the field of immunotherapy, across a variety of commonly used gene panels. Regardless of the panel, we observed improvements in regression to the gold standard whole exome derived value for this metric, with additional performance benefits as more data were provided to the model (such as noncoding variants from panel assays). Our results suggest this framework could lead to improvements in a range of tasks where the sample level metric is determined by the aggregation of a set of genomic measures, such as somatic mutations that we focused on in this study.