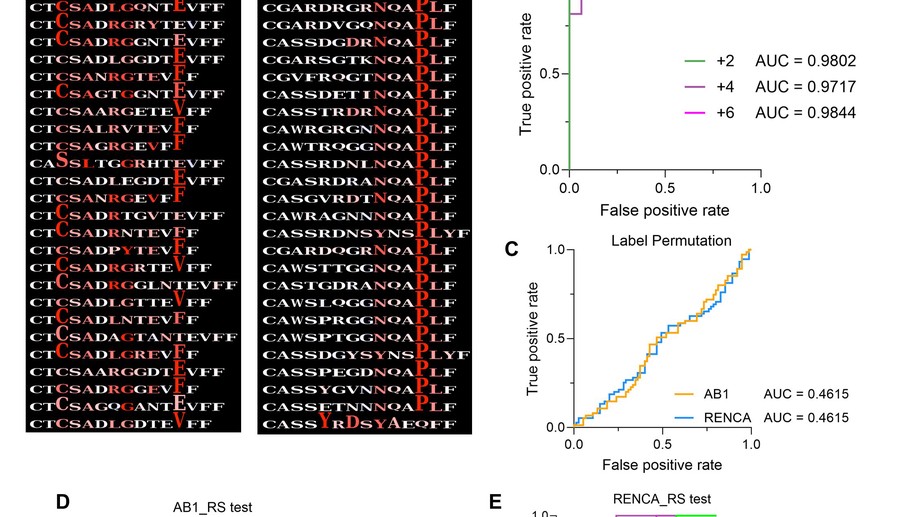
Immune checkpoint therapy responders display early clonal expansion of tumor infiltrating lymphocytes
Immune checkpoint therapy (ICT) causes durable tumour responses in a subgroup of patients, but it is not well known how T cell receptor beta (TCRβ) repertoire dynamics contribute to the therapeutic response. Using murine models that exclude variation in host genetics, environmental factors and tumour mutation burden, limiting variation between animals to naturally diverse TCRβ repertoires, we applied TCRseq, single cell RNAseq and flow cytometry to study TCRβ repertoire dynamics in ICT responders and non-responders. Increased oligoclonal expansion of TCRβ clonotypes was observed in responding tumours. Machine learning identified TCRβ CDR3 signatures unique to each tumour model, and signatures associated with ICT response at various timepoints before or during ICT. Clonally expanded CD8+ T cells in responding tumours post ICT displayed effector T cell gene signatures and phenotype. An early burst of clonal expansion during ICT is associated with response, and we report unique dynamics in TCRβ signatures associated with ICT response.
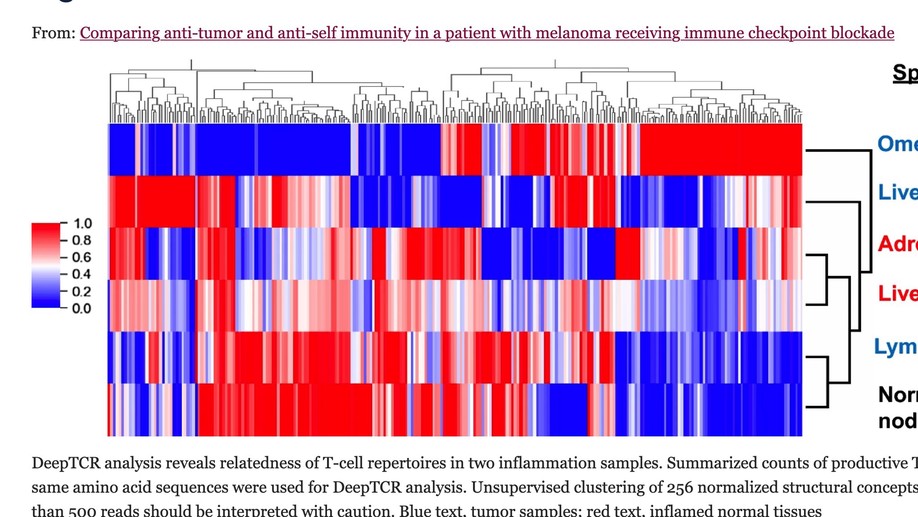
Comparing anti-tumor and anti-self immunity in a patient with melanoma receiving immune checkpoint blockade
Tumor regression following immune checkpoint blockade (ICB) is often associated with immune-related adverse events (irAEs), marked by inflammation in non-cancerous tissues. This study was undertaken to investigate the functional relationship between anti-tumor and anti-self immunity, to facilitate irAE management while promoting anti-tumor immunity. Multiple biopsies from tumor and inflamed tissues were collected from a patient with melanoma experiencing both tumor regression and irAEs on ICB, who underwent rapid autopsy. Immune cells infiltrating melanoma lesions and inflamed normal tissues were subjected to gene expression profiling with multiplex qRT-PCR for 122 candidate genes. Subsequently, immunohistochemistry was conducted to assess the expression of 14 candidate markers of immune cell subsets and checkpoints. TCR-beta sequencing was used to explore T cell clonal repertoires across specimens. While genes involved in MHC I/II antigen presentation, IFN signaling, innate immunity and immunosuppression were abundantly expressed across specimens, irAE tissues over-expressed certain genes associated with immunosuppression (CSF1R, IL10RA, IL27/EBI3, FOXP3, KLRG1, SOCS1, TGFB1), including those in the COX-2/PGE2 pathway (IL1B, PTGER1/EP1 and PTGER4/EP4). Immunohistochemistry revealed similar proportions of immunosuppressive cell subsets and checkpoint molecules across samples. TCRseq did not indicate common TCR repertoires across tumor and inflammation sites, arguing against shared antigen recognition between anti-tumor and anti-self immunity in this patient. This comprehensive study of a single patient with melanoma experiencing both tumor regression and irAEs on ICB explores the immune landscape across these tissues, revealing similarities between anti-tumor and anti-self immunity. Further, it highlights expression of the COX-2/PGE2 pathway, which is known to be immunosuppressive and potentially mediates ICB resistance. Ongoing clinical trials of COX-2/PGE2 pathway inhibitors targeting the major COX-2 inducer IL-1B, COX-2 itself, or the PGE2 receptors EP2 and EP4 present new opportunities to promote anti-tumor activity, but may also have the potential to enhance the severity of ICB-induced irAEs.
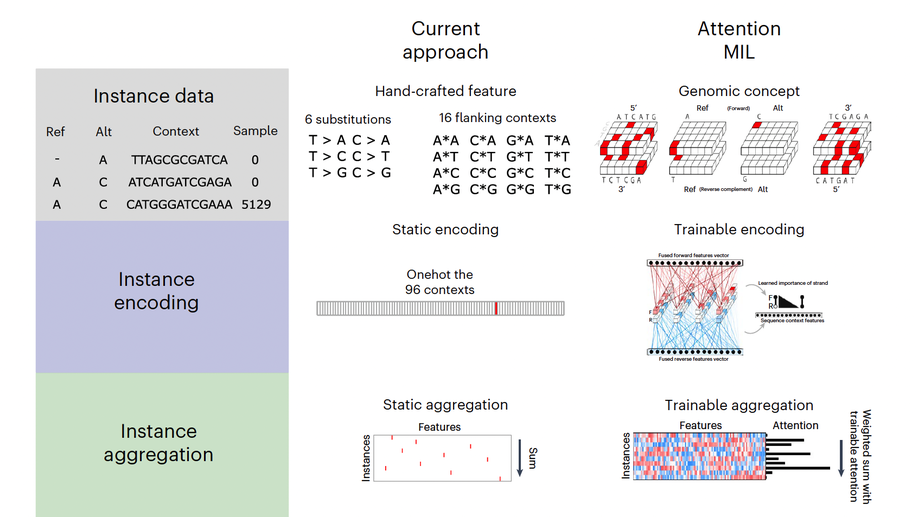
Multiple-instance learning of somatic mutations for the classification of tumour type and the prediction of microsatellite status
Large-scale genomic data are well suited to analysis by deep learning algorithms. However, for many genomic datasets, labels are at the level of the sample rather than for individual genomic measures. Machine learning models leveraging these datasets generate predictions by using statically encoded measures that are then aggregated at the sample level. Here we show that a single weakly supervised end-to-end multiple-instance-learning model with multi-headed attention can be trained to encode and aggregate the local sequence context or genomic position of somatic mutations, hence allowing for the modelling of the importance of individual measures for sample-level classification and thus providing enhanced explainability. The model solves synthetic tasks that conventional models fail at, and achieves best-in-class performance for the classification of tumour type and for predicting microsatellite status. By improving the performance of tasks that require aggregate information from genomic datasets, multiple-instance deep learning may generate biological insight.
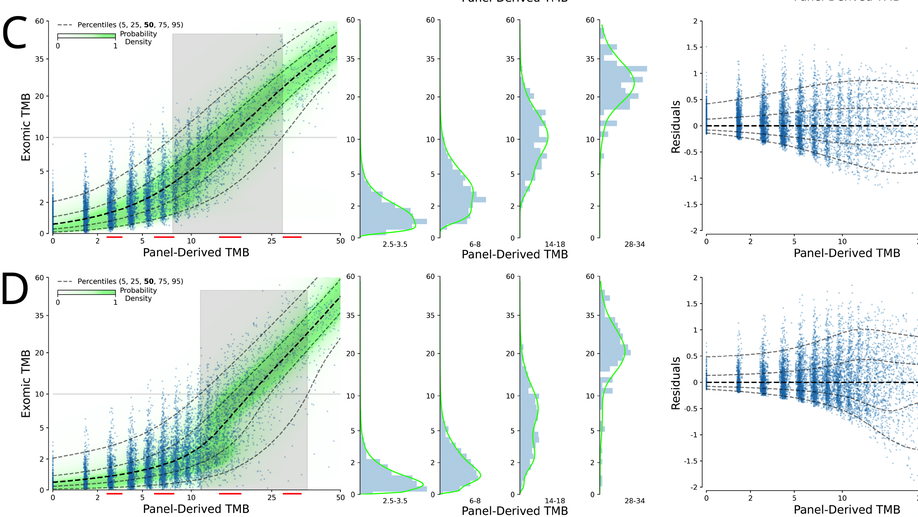
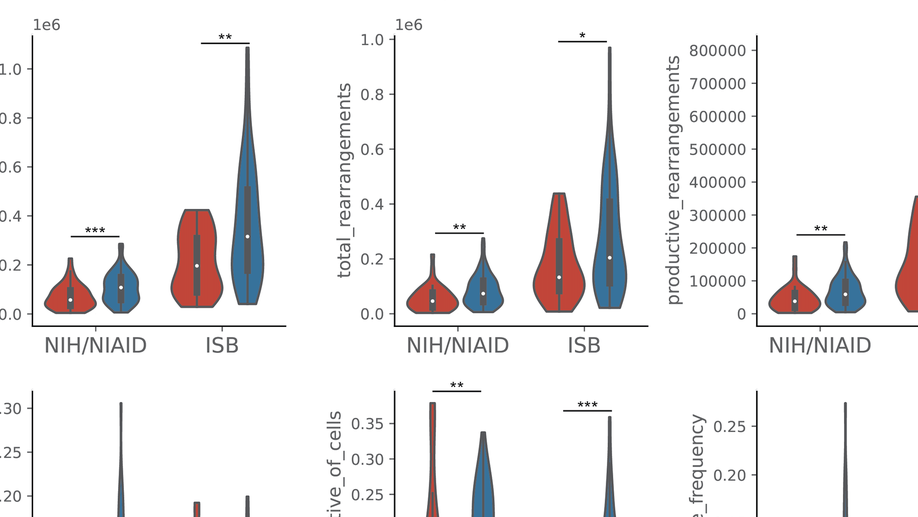
Probabilistic mixture models improve calibration of panel-derived tumor mutational burden in the context of both tumor-normal and tumor-only sequencing
Background: Tumor mutational burden (TMB) has been investigated as a biomarker for immune checkpoint blockade (ICB) therapy. Increasingly, TMB is being estimated with gene panel-based assays (as opposed to full exome sequencing) and different gene panels cover overlapping but distinct genomic coordinates, making comparisons across panels difficult. Previous studies have suggested that standardization and calibration to exome-derived TMB be done for each panel to ensure comparability. With TMB cutoffs being developed from panel-based assays, there is a need to understand how to properly estimate exomic TMB values from different panel-based assays. Design: Our approach to calibration of panel-derived TMB to exomic TMB proposes the use of probabilistic mixture models that allow for nonlinear relationships along with heteroscedastic error. We examined various inputs including nonsynonymous, synonymous, and hotspot counts along with genetic ancestry. Using the TCGA cohort we generated a tumor-only version of the panel-restricted data by reintroducing private germline variants. Results: We were able to model more accurately the distribution of both tumor-normal and tumor-only data using the proposed probabilistic mixture models as compared to linear regression. Applying a model trained on tumor-normal data to tumor-only input results in biased TMB predictions. Including synonymous mutations resulted in better regression metrics across both data types, but ultimately a model able to dynamically weight the various input mutation types exhibited optimal performance. Including genetic ancestry improved model performance only in the context of tumor-only data, wherein private germline variants are observed.
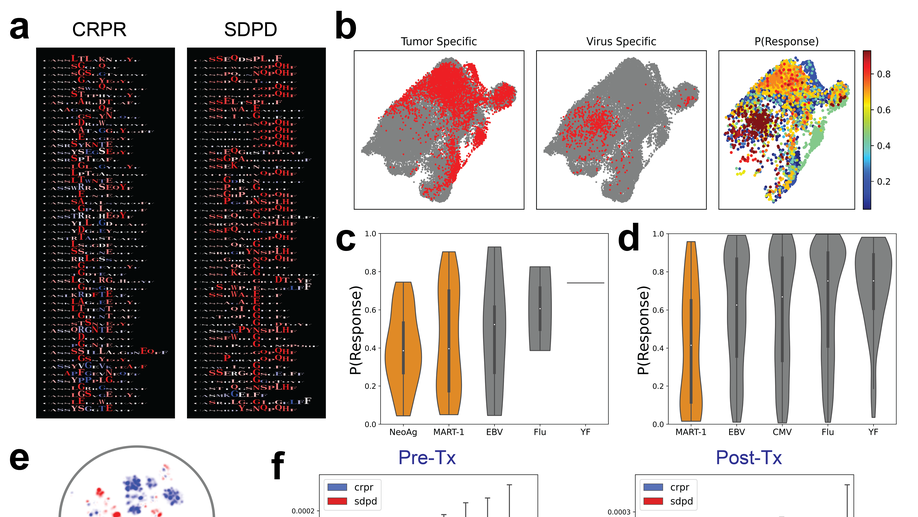
Deep learning reveals predictive sequence concepts within immune repertoires to immunotherapy
T cell receptor (TCR) sequencing has been used to characterize the immune response to cancer. However, most analyses have been restricted to quantitative measures such as clonality that do not leverage the complementarity-determining region 3 (CDR3) sequence. We use DeepTCR, a framework of deep learning algorithms, to reveal sequence concepts that are predictive of response to immunotherapy. We demonstrate that DeepTCR can predict response and use the model to infer the antigenic specificities of the predictive signature and their unique dynamics during therapy. The predictive signature of nonresponse is associated with high frequencies of TCRs predicted to recognize tumor-specific antigens, and these tumor-specific TCRs undergo a higher degree of dynamic changes on therapy in nonresponders versus responders. These results are consistent with a biological model where the hallmark of nonresponders is an accumulation of tumor-specific T cells that undergo turnover on therapy, possibly because of the dysfunctional state of these T cells in nonresponders.
Deep learning identifies antigenic determinants of severe SARS-CoV-2 infection within T-cell repertoires
SARS-CoV-2 infection is characterized by a highly variable clinical course with patients experiencing asymptomatic infection all the way to requiring critical care support. This variation in clinical course has led physicians and scientists to study factors that may predispose certain individuals to more severe clinical presentations in hopes of either identifying these individuals early in their illness or improving their medical management. We sought to understand immunogenomic differences that may result in varied clinical outcomes through analysis of T-cell receptor sequencing (TCR-Seq) data in the open access ImmuneCODE database. We identified two cohorts within the database that had clinical outcomes data reflecting severity of illness and utilized DeepTCR, a multiple-instance deep learning repertoire classifier, to predict patients with severe SARS-CoV-2 infection from their repertoire sequencing. We demonstrate that patients with severe infection have repertoires with higher T-cell responses associated with SARS-CoV-2 epitopes and identify the epitopes that result in these responses. Our results provide evidence that the highly variable clinical course seen in SARS-CoV-2 infection is associated to certain antigen-specific responses.
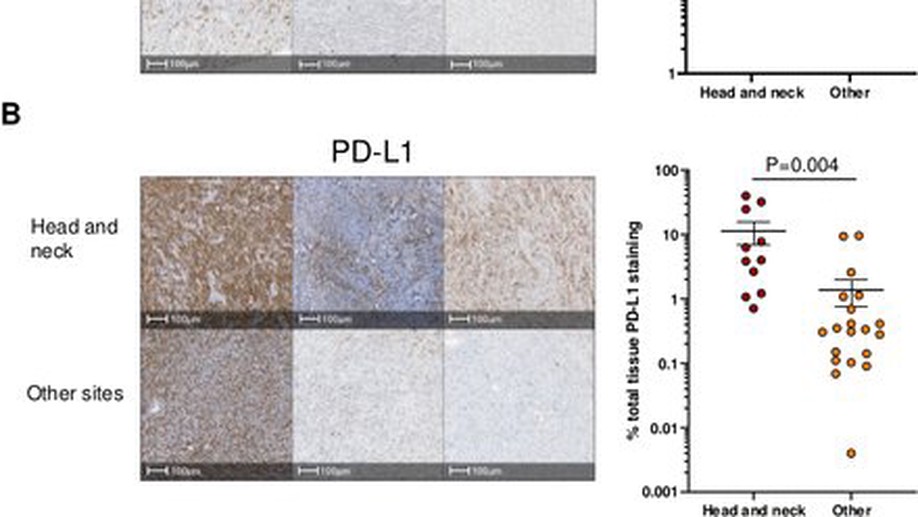
Anti-PD-1 elicits regression of undifferentiated pleomorphic sarcomas with UV-mutation signatures
textlessptextgreaterUndifferentiated pleomorphic sarcoma (UPS), an aggressive soft-tissue sarcoma of adults, has been characterized by low tumor mutational burden (TMB) and high copy number alterations. Clinical trials of programmed death-1 (PD-1) blockade in UPS have reported widely varying efficacy. We describe two patients with recurrent scalp UPS that experienced clinical benefit from PD-1 blockade. These tumors had high TMB with a UV-induced mutational pattern. Analysis of additional head and neck UPS cases identified five out of seven tumors with high TMB and an ultraviolet (UV) mutational signature. Head and neck UPS tumors also had increased programmed death-ligand 1 (PD-L1) expression and CD8+ T cell infiltration as compared with UPS tumors arising from other sites. In summary, we found that UPS tumors of the head and neck, but not elsewhere, have a PD-L1+, T-cell-inflamed tumor microenvironment and high TMB, suggesting that these tumors represent a distinct genetic subgroup of UPS for which immune checkpoint inhibitor therapy might be effective.textless/ptextgreater
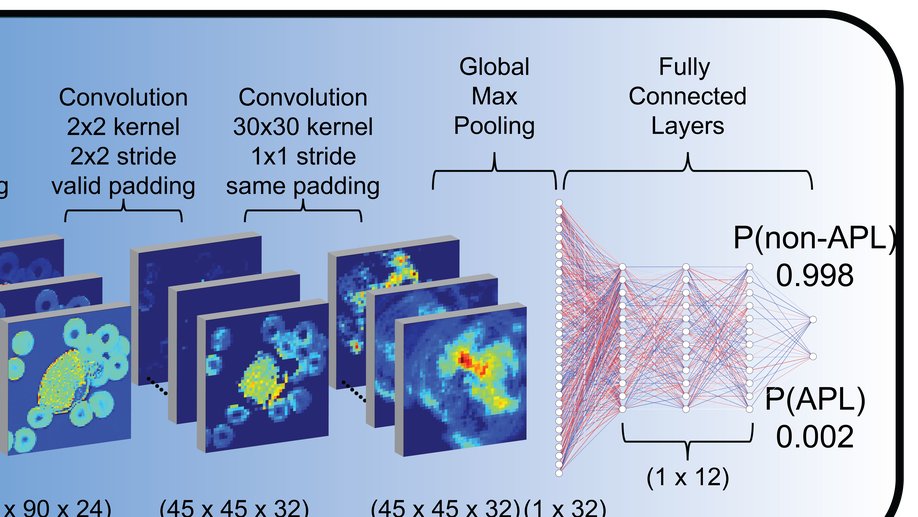
Deep learning for diagnosis of acute promyelocytic leukemia via recognition of genomically imprinted morphologic features
Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia (AML), classified by a translocation between chromosomes 15 and 17 [t(15;17)], that is considered a true oncologic emergency though appropriate therapy is considered curative. Therapy is often initiated on clinical suspicion, informed by both clinical presentation as well as direct visualization of the peripheral smear. We hypothesized that genomic imprinting of morphologic features learned by deep learning pattern recognition would have greater discriminatory power and consistency compared to humans, thereby facilitating identification of t(15;17) positive APL. By applying both cell-level and patient-level classification linked to t(15;17) PML/RARA ground-truth, we demonstrate that deep learning is capable of distinguishing APL in both discovery and prospective independent cohort of patients. Furthermore, we extract learned information from the trained network to identify previously undescribed morphological features of APL. The deep learning method we describe herein potentially allows a rapid, explainable, and accurate physician-aid for diagnosing APL at the time of presentation in any resource-poor or -rich medical setting given the universally available peripheral smear.
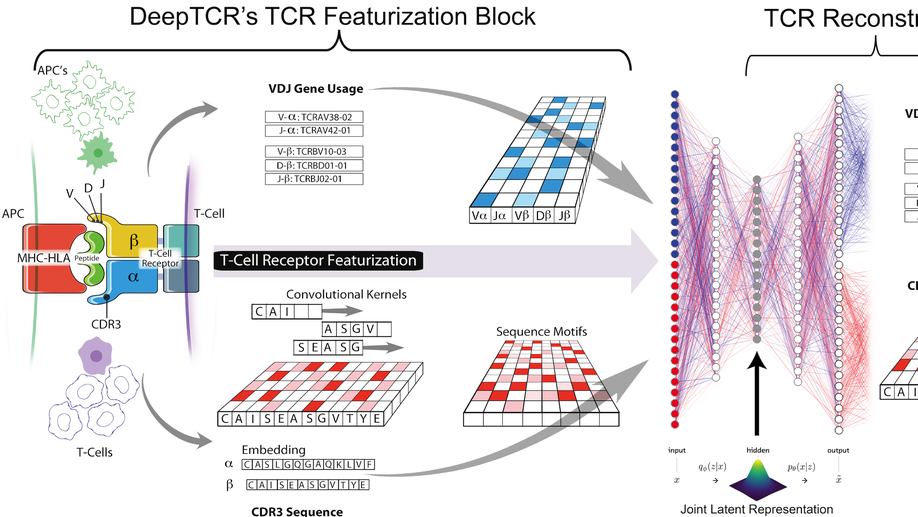
DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires
Deep learning algorithms have been utilized to achieve enhanced performance in pattern-recognition tasks. The ability to learn complex patterns in data has tremendous implications in immunogenomics. T-cell receptor (TCR) sequencing assesses the diversity of the adaptive immune system and allows for modeling its sequence determinants of antigenicity. We present DeepTCR, a suite of unsupervised and supervised deep learning methods able to model highly complex TCR sequencing data by learning a joint representation of a TCR by its CDR3 sequences and V/D/J gene usage. We demonstrate the utility of deep learning to provide an improved ‘featurization’ of the TCR across multiple human and murine datasets, including improved classification of antigen-specific TCRs and extraction of antigen-specific TCRs from noisy single-cell RNA-Seq and T-cell culture-based assays. Our results highlight the flexibility and capacity for deep neural networks to extract meaningful information from complex immunogenomic data for both descriptive and predictive purposes.
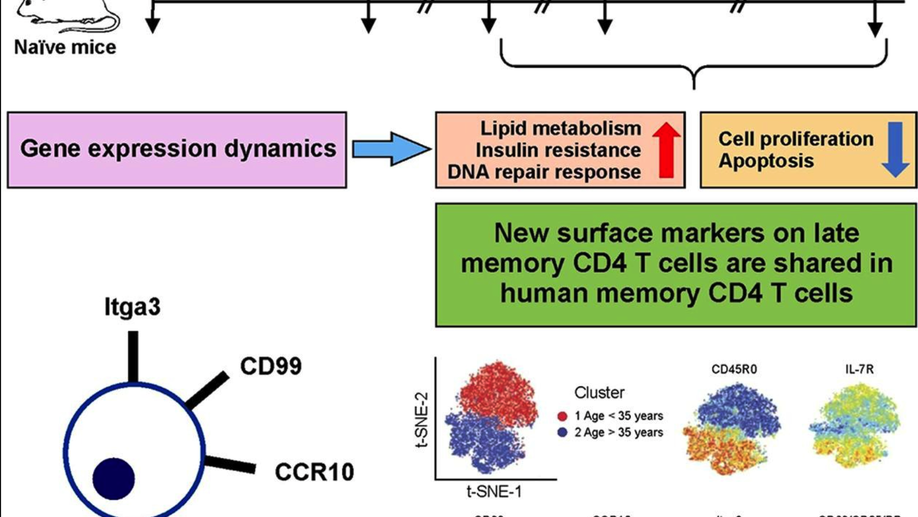
Multiple genetic programs contribute to CD4 T cell memory differentiation and longevity by maintaining T cell quiescence
While memory T-cells represent a hallmark of adaptive immunity, little is known about the genetic mechanisms regulating the longevity of memory CD4 T cells. Here, we studied the dynamics of gene expression in antigen specific CD4 T cells during infection, memory differentiation, and long-term survival up to nearly a year in mice. We observed that differentiation into long lived memory cells is associated with increased expression of genes inhibiting cell proliferation and apoptosis as well as genes promoting DNA repair response, lipid metabolism, and insulin resistance. We identified several transmembrane proteins in long-lived murine memory CD4 T cells, which co-localized exclusively within the responding antigen-specific memory CD4 T cells in human. The unique gene signatures of long-lived memory CD4 T cells, along with the new markers that we have defined, will enable a deeper understanding of memory CD4 T cell biology and allow for designing novel vaccines and therapeutics.
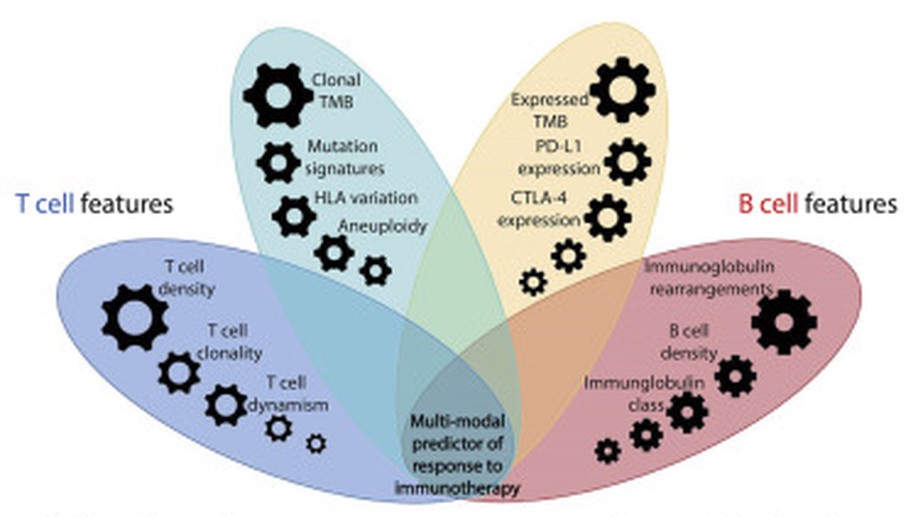
Integrative Tumor and Immune Cell Multi-omic Analyses Predict Response to Immune Checkpoint Blockade in Melanoma
In this study, we incorporate analyses of genome-wide sequence and structural alterations with pre- and on-therapy transcriptomic and T cell repertoire features in immunotherapy-naive melanoma patients treated with immune checkpoint blockade. Although tumor mutation burden is associated with improved treatment response, the mutation frequency in expressed genes is superior in predicting outcome. Increased T cell density in baseline tumors and dynamic changes in regression or expansion of the T cell repertoire during therapy distinguish responders from non-responders. Transcriptome analyses reveal an increased abundance of B cell subsets in tumors from responders and patterns of molecular response related to expressed mutation elimination or retention that reflect clinical outcome. High-dimensional genomic, transcriptomic, and immune repertoire data were integrated into a multi-modal predictor of response. These findings identify genomic and transcriptomic characteristics of tumors and immune cells that predict response to immune checkpoint blockade and highlight the importance of pre-existing T and B cell immunity in therapeutic outcomes.
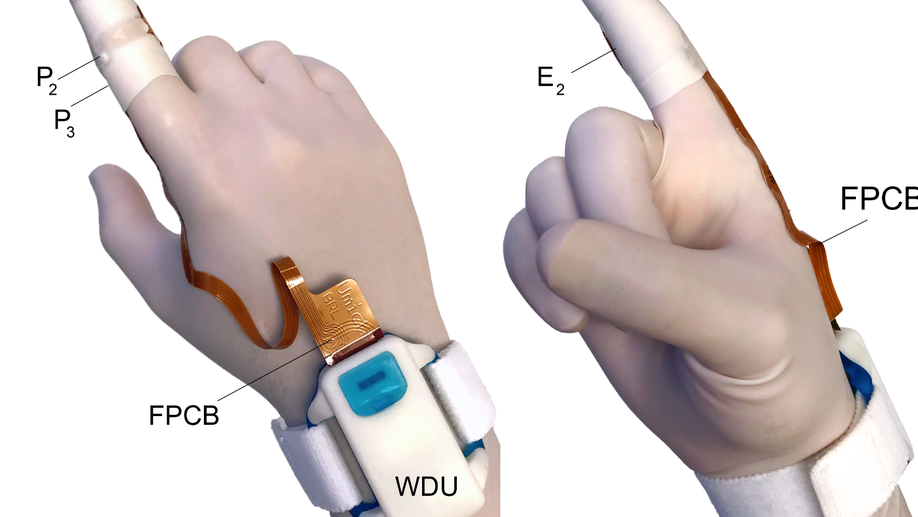
Comparison of anorectal function measured using wearable digital manometry and a high resolution manometry system
There is a need for a lower cost manometry system for assessing anorectal function in primary and secondary care settings. We developed an index finger-based system (termed “digital manometry”) and tested it in healthy volunteers, patients with chronic constipation, and fecal incontinence. Anorectal pressures were measured in 16 participants with the digital manometry system and a 23-channel high-resolution anorectal manometry system. The results were compared using a Bland-Altman analysis at rest as well as during maximum squeeze and simulated defecation maneuvers. Myoelectric activity of the puborectalis muscle was also quantified simultaneously using the digital manometry system. The limits of agreement between the two methods were -7.1 ± 25.7 mmHg for anal sphincter resting pressure, 0.4 ± 23.0 mmHg for the anal sphincter pressure change during simulated defecation, -37.6 ± 50.9 mmHg for rectal pressure changes during simulated defecation, and -20.6 ± 172.6 mmHg for anal sphincter pressure during the maximum squeeze maneuver. The change in the puborectalis myoelectric activity was proportional to the anal sphincter pressure increment during a maximum squeeze maneuver (slope = 0.6, R2 = 0.4). Digital manometry provided a similar evaluation of anorectal pressures and puborectalis myoelectric activity at an order of magnitude less cost than high-resolution manometry, and with a similar level of patient comfort. Digital Manometry provides a simple, inexpensive, point of service means of assessing anorectal function in patients with chronic constipation and fecal incontinence.
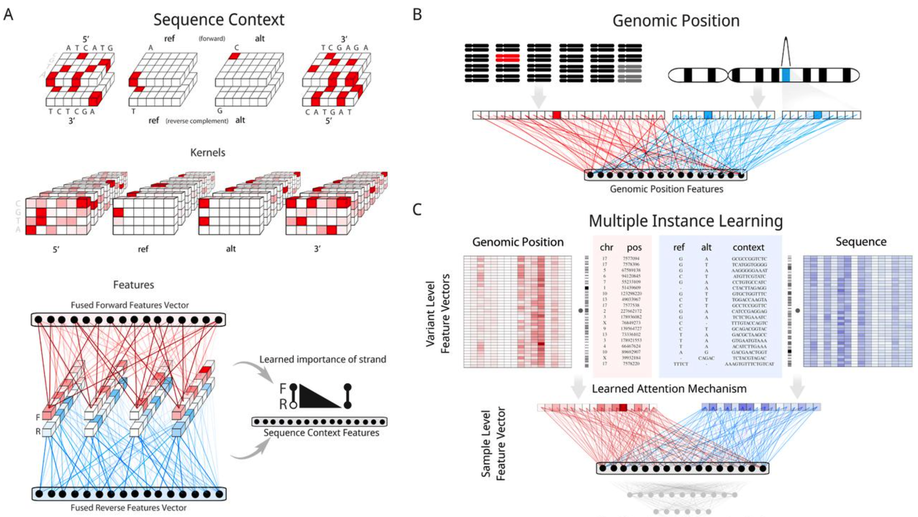
Aggregation Tool for Genomic Concepts (ATGC): A deep learning framework for sparse genomic measures and its application to tumor mutational burden
Deep learning has the ability to extract meaningful features from data given enough training examples. Large scale genomic data are well suited for this class of machine learning algorithms; however, for many of these data the labels are at the level of the sample instead of at the level of the individual genomic measures. To leverage the power of deep learning for these types of data we turn to a multiple instance learning framework, and present an easily extensible tool built with TensorFlow and Keras. We show how this tool can be applied to somatic variants (featurizing genomic position and sequence context), and accurately classify samples according to whether they contain a specific variant (hotspot or tumor suppressor) or whether they contain a type of variant (microsatellite instability). We then apply our model to the calibration of tumor mutational burden (TMB), an increasingly important metric in the field of immunotherapy, across a variety of commonly used gene panels. Regardless of the panel, we observed improvements in regression to the gold standard whole exome derived value for this metric, with additional performance benefits as more data were provided to the model (such as noncoding variants from panel assays). Our results suggest this framework could lead to improvements in a range of tasks where the sample level metric is determined by the aggregation of a set of genomic measures, such as somatic mutations that we focused on in this study.
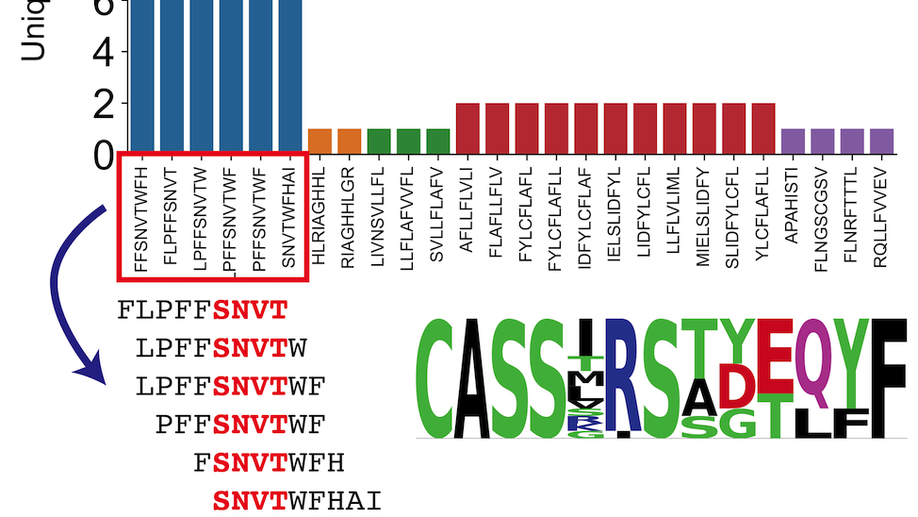
Analysis of SARS-CoV-2 specific T-cell receptors in ImmuneCode reveals cross-reactivity to immunodominant Influenza M1 epitope
Adaptive Biotechnologies and Microsoft have recently partnered to release ImmuneCode, a database containing SARS-CoV-2 specific T-cell receptors derived through MIRA, a T-cell receptor (TCR) sequencing based sequencing approach to identify antigen-specific TCRs. Herein, we query the extent of cross reactivity between these derived SARS-CoV-2 specific TCRs and other known antigens present in McPas-TCR, a manually curated catalogue of pathology-associated TCRs. We reveal cross reactivity between SARS-CoV-2 specific TCRs and the immunodominant Influenza GILGFVFTL M1 epitope, suggesting the importance of further work in characterizing the implications of prior Influenza exposure or co-exposure to the pathology of SARS-CoV-2 illness.
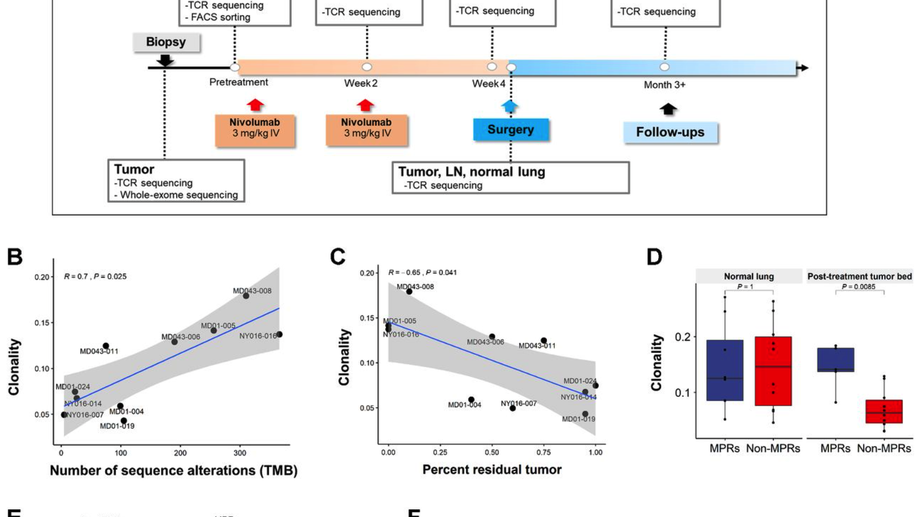
Compartmental Analysis of T-cell Clonal Dynamics as a Function of Pathologic Response to Neoadjuvant PD-1 Blockade in Resectable Non–Small Cell Lung Cancer
Purpose: Neoadjuvant PD-1 blockade is a promising treatment for resectable non–small cell lung cancer (NSCLC), yet immunologic mechanisms contributing to tumor regression and biomarkers of response are unknown. Using paired tumor/blood samples from a phase II clinical trial (NCT02259621), we explored whether the peripheral T-cell clonotypic dynamics can serve as a biomarker for response to neoadjuvant PD-1 blockade. Experimental Design: T-cell receptor (TCR) sequencing was performed on serial peripheral blood, tumor, and normal lung samples from resectable NSCLC patients treated with neoadjuvant PD-1 blockade. We explored the temporal dynamics of the T-cell repertoire in the peripheral and tumoral compartments in response to neoadjuvant PD-1 blockade by using the TCR as a molecular barcode. Results: Higher intratumoral TCR clonality was associated with reduced percent residual tumor at the time of surgery, and the TCR repertoire of tumors with major pathologic response (MPR; textless10% residual tumor after neoadjuvant therapy) had a higher clonality and greater sharing of tumor-infiltrating clonotypes with the peripheral blood relative to tumors without MPR. Additionally, the posttreatment tumor bed of patients with MPR was enriched with T-cell clones that had peripherally expanded between weeks 2 and 4 after anti–PD-1 initiation and the intratumoral space occupied by these clonotypes was inversely correlated with percent residual tumor. Conclusions: Our study suggests that exchange of T-cell clones between tumor and blood represents a key correlate of pathologic response to neoadjuvant immunotherapy and shows that the periphery may be a previously underappreciated originating compartment for effective antitumor immunity.See related commentary by Henick, p. 1205
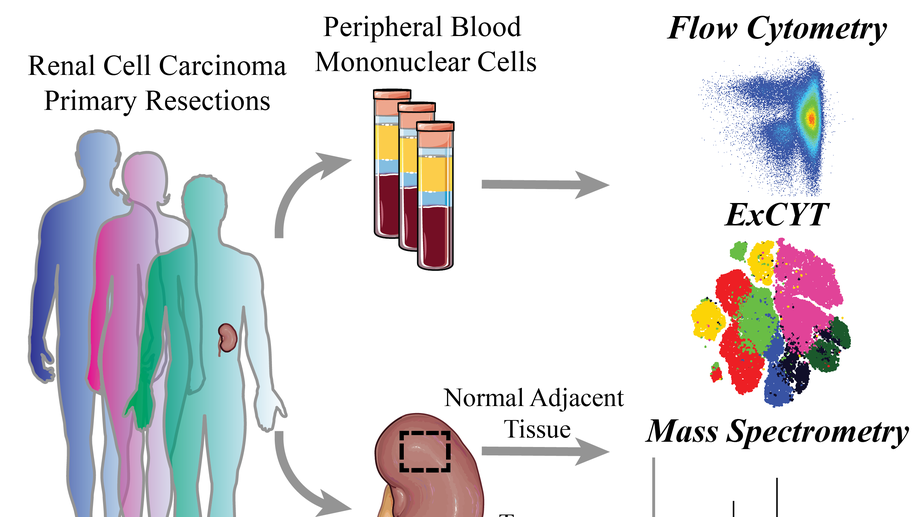
High-Dimensional Cytometry (ExCYT) and Mass Spectrometry of myeloid infiltrate in clinically localized clear cell Renal Cell Carcinoma identifies novel potential myeloid targets for immunotherapy
Renal Cell Carcinoma (RCC) is one of the most commonly diagnosed cancers worldwide with research efforts dramatically improving understanding of the biology of the disease. To investigate the role of the immune system in treatment-naïve clear cell Renal Cell Carcinoma (ccRCC), we interrogated the immune infiltrate in patient-matched ccRCC tumor samples, benign normal adjacent tissue (NAT) and peripheral blood mononuclear cells (PBMCs isolated from whole blood, focusing our attention on the myeloid cell infiltrate. Using flow cytometric, mass spectrometry, and ExCYT analysis, we discovered unique myeloid populations in PBMCs across patient samples. Furthermore, normal adjacent tissues and ccRCC tissues contained numerous myeloid populations with a unique signature for both tissues. Enrichment of the immune cell (CD45+) fraction and subsequent gene expression analysis revealed a number of myeloid-related genes that were differentially expressed. These data provide evidence, for the first time, of an immunosuppressive and pro-tumorigenic role of myeloid cells in early, clinically localized ccRCC. The identification of a number of immune proteins for therapeutic targeting provides a rationale for investigation into the potential efficacy of earlier intervention with single-agent or combination immunotherapy for ccRCC.
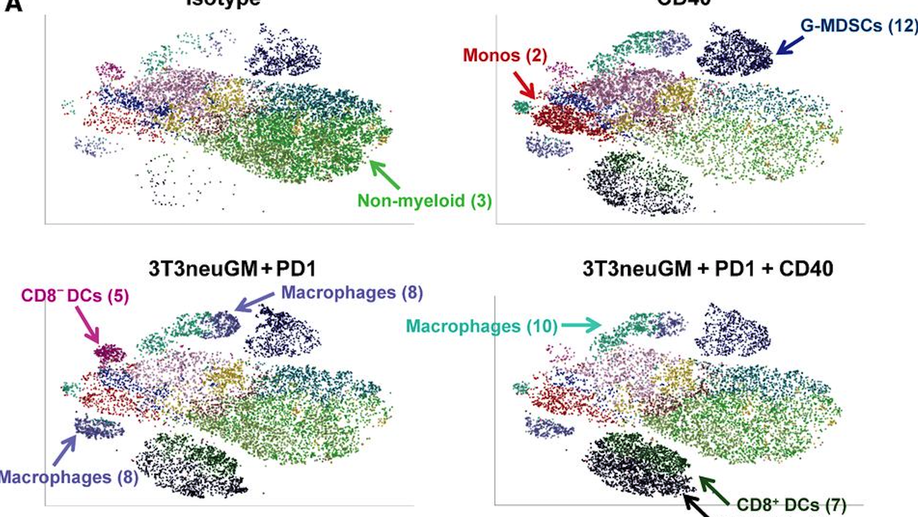
A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell–Mediated Anticancer Activity
In cancers with tumor-infiltrating lymphocytes (TILs), monoclonal antibodies (mAbs) that block immune checkpoints such as CTLA-4 and PD-1/PD-L1 promote antitumor T-cell immunity. Unfortunately, most cancers fail to respond to single-agent immunotherapies. T regulatory cells, myeloid derived suppressor cells (MDSCs), and extensive stromal networks within the tumor microenvironment (TME) dampen antitumor immune responses by preventing T-cell infiltration and/or activation. Few studies have explored combinations of immune-checkpoint antibodies that target multiple suppressive cell populations within the TME, and fewer have studied the combinations of both agonist and antagonist mAbs on changes within the TME. Here, we test the hypothesis that combining a T-cell–inducing vaccine with both a PD-1 antagonist and CD40 agonist mAbs (triple therapy) will induce T-cell priming and TIL activation in mouse models of nonimmunogenic solid malignancies. In an orthotopic breast cancer model and both subcutaneous and metastatic pancreatic cancer mouse models, only triple therapy was able to eradicate most tumors. The survival benefit was accompanied by significant tumor infiltration of IFNγ-, Granzyme B-, and TNFα-secreting effector T cells. Further characterization of immune populations was carried out by high-dimensional flow-cytometric clustering analysis and visualized by t-distributed stochastic neighbor embedding (t-SNE). Triple therapy also resulted in increased infiltration of dendritic cells, maturation of antigen-presenting cells, and a significant decrease in granulocytic MDSCs. These studies reveal that combination CD40 agonist and PD-1 antagonist mAbs reprogram immune resistant tumors in favor of antitumor immunity.
Persistent mutant oncogene specific T cells in two patients benefitting from anti-PD-1
Several predictive biomarkers are currently approved or are under investigation for the selection of patients for checkpoint blockade. Tumor PD-L1 expression is used for stratification of non-small cell lung (NSCLC) patients, with tumor mutational burden (TMB) also being explored with promising results, and mismatch-repair deficiency is approved for tumor site-agnostic disease. While tumors with high PD-L1 expression, high TMB, or mismatch repair deficiency respond well to checkpoint blockade, tumors with lower PD-L1 expression, lower mutational burdens, or mismatch repair proficiency respond much less frequently.
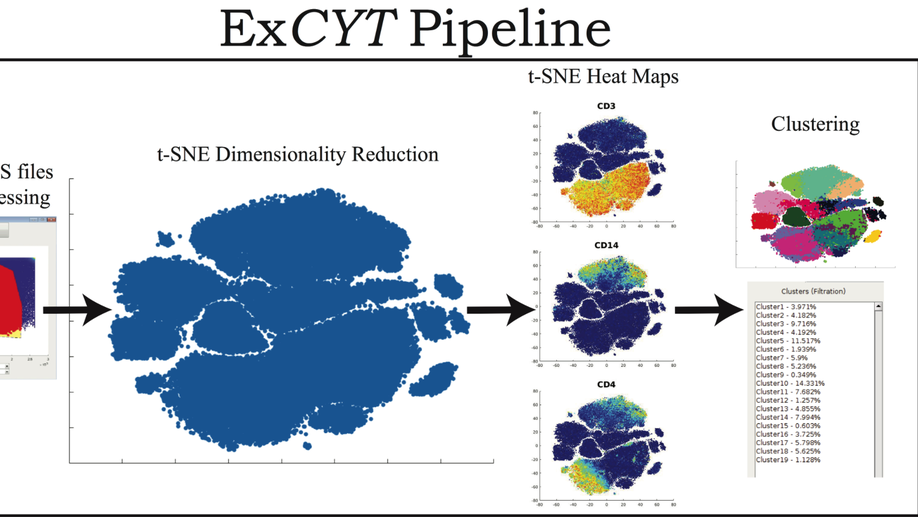
ExCYT: A Graphical User Interface for Streamlining Analysis of High-Dimensional Cytometry Data
With the advent of flow cytometers capable of measuring an increasing number of parameters, scientists continue to develop larger panels to phenotypically explore characteristics of their cellular samples. However, these technological advancements yield high-dimensional data sets that have become increasingly difficult to analyze objectively within traditional manual-based gating programs. In order to better analyze and present data, scientists partner with bioinformaticians with expertise in analyzing high-dimensional data to parse their flow cytometry data. While these methods have been shown to be highly valuable in studying flow cytometry, they have yet to be incorporated in a straightforward and easy-to-use package for scientists who lack computational or programming expertise. To address this need, we have developed ExCYT, a MATLAB-based Graphical User Interface (GUI) that streamlines the analysis of high-dimensional flow cytometry data by implementing commonly employed analytical techniques for high-dimensional data including dimensionality reduction by t-SNE, a variety of automated and manual clustering methods, heatmaps, and novel high-dimensional flow plots. Additionally, ExCYT provides traditional gating options of select populations of interest for further t-SNE and clustering analysis as well as the ability to apply gates directly on t-SNE plots. The software provides the additional advantage of working with either compensated or uncompensated FCS files. In the event that post-acquisition compensation is required, the user can choose to provide the program a directory of single stains and an unstained sample. The program detects positive events in all channels and uses this select data to more objectively calculate the compensation matrix. In summary, ExCYT provides a comprehensive analysis pipeline to take flow cytometry data in the form of FCS files and allow any individual, regardless of computational training, to use the latest algorithmic approaches in understanding their data.
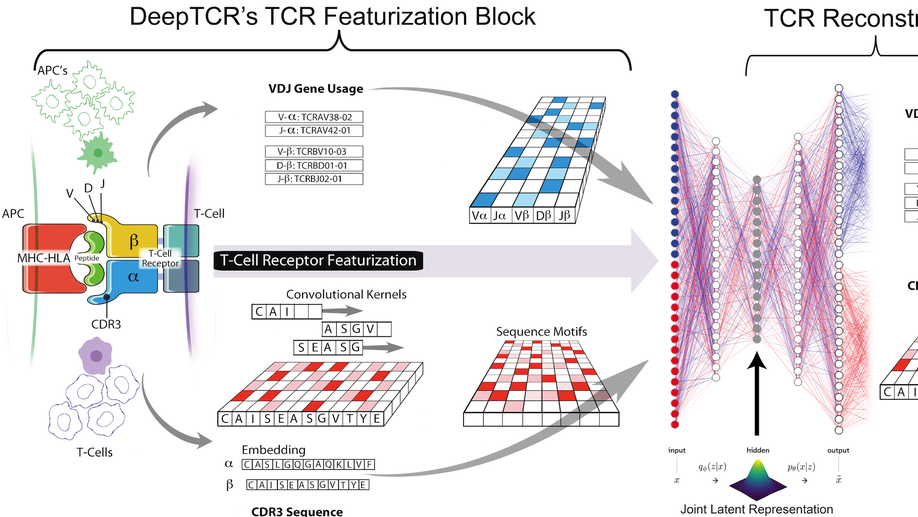
DeepTCR: a deep learning framework for revealing structural concepts within TCR Repertoire
Deep learning algorithms have been utilized to achieve excellent performance in pattern-recognition tasks, such as in image and vocal recognition$^textrm1,2$. The ability to learn complex patterns in data has tremendous implications in the genomics world, where sequence motifs become learned ‘features’ that can be used to predict functionality, guiding our understanding of disease and basic biology$^textrm3–6$. T-cell receptor (TCR) sequencing assesses the diversity of the adaptive immune system, and while prior conventional biological sequence analysis tools have been insightful, they have significant shortcomings. Prior approaches have been limited to single-sequence analytics and thus unable to characterize the overall structural information within an entire sample of sequences. Furthermore, they utilize only unsupervised approaches, being unable to leverage labels to guide the learning process$^textrm7–9$. We present DeepTCR, a broad collection of unsupervised and supervised deep learning methods able to uncover structure in highly complex and large TCR sequencing data. We demonstrate its utility across multiple scientific examples, including learning antigen-specific motifs to viral and tumor-specific epitopes and understanding immunotherapy-related shaping of repertoire. We further extract meaningful motifs from the trained network as a means of explaining the sequence concepts that have been learned to accomplish a given task. Finally, we extend DeepTCR’s functionality to analyze paired α/β chains as inputs, demonstrating the ability to query the contribution of each chain to antigen-specificity. Our results show the flexibility and capacity for deep neural networks to handle the complexity of high-dimensional genomics data for both descriptive and predictive purposes.
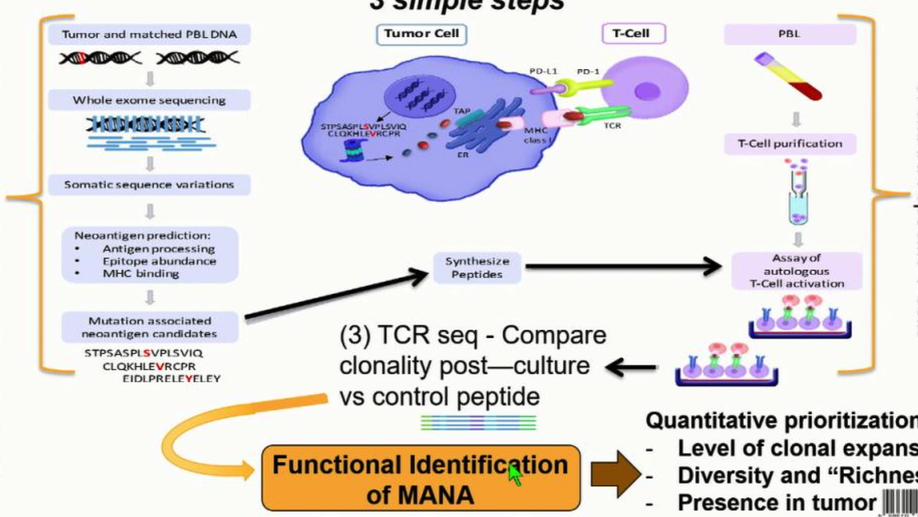
The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity
Mutation-associated neoantigens (MANA) are a target of antitumor T-cell immunity. Sensitive, simple, and standardized assays are needed to assess the repertoire of functional MANA-specific T cells in oncology. Assays analyzing in vitro cytokine production such as ELISpot and intracellular cytokine staining have been useful but have limited sensitivity in assessing tumor-specific T-cell responses and do not analyze antigen-specific T-cell repertoires. The FEST (Functional Expansion of Specific T cells) assay described herein integrates T-cell receptor sequencing of short-term, peptide-stimulated cultures with a bioinformatic platform to identify antigen-specific clonotypic amplifications. This assay can be adapted for all types of antigens, including MANAs via tumor exome-guided prediction of MANAs. Following in vitro identification by the MANAFEST assay, the MANA-specific CDR3 sequence can be used as a molecular barcode to detect and monitor the dynamics of these clonotypes in blood, tumor, and normal tissue of patients receiving immunotherapy. MANAFEST is compatible with high-throughput routine clinical and lab practices. Cancer Immunol Res; 6(8); 888–99. ©2018 AACR.
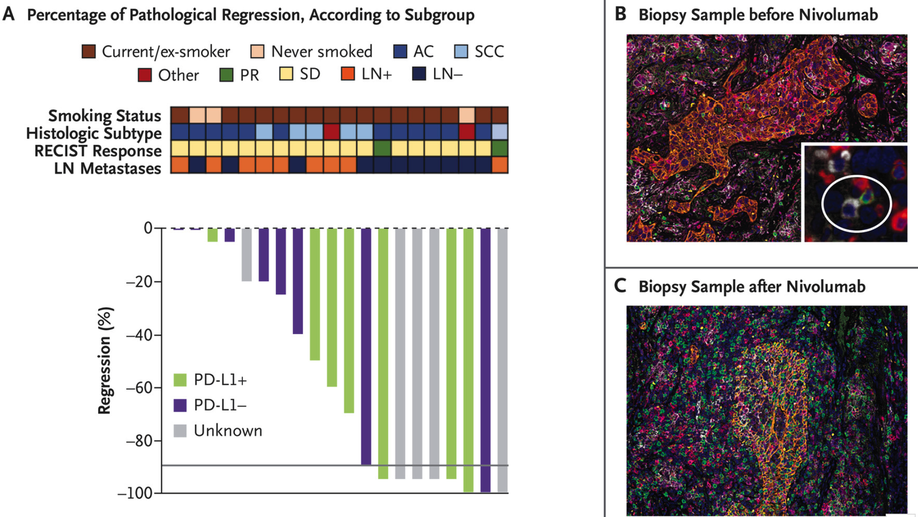
Neoadjuvant PD-1 Blockade in Resectable Lung Cancer
Neoadjuvant PD-1 Blockade in Lung Cancer In a pilot study, two doses of neoadjuvant nivolumab administered to patients with resectable lung cancer resulted in a major pathological response in 45% and amplified T-cell clones specific for tumor antigens.
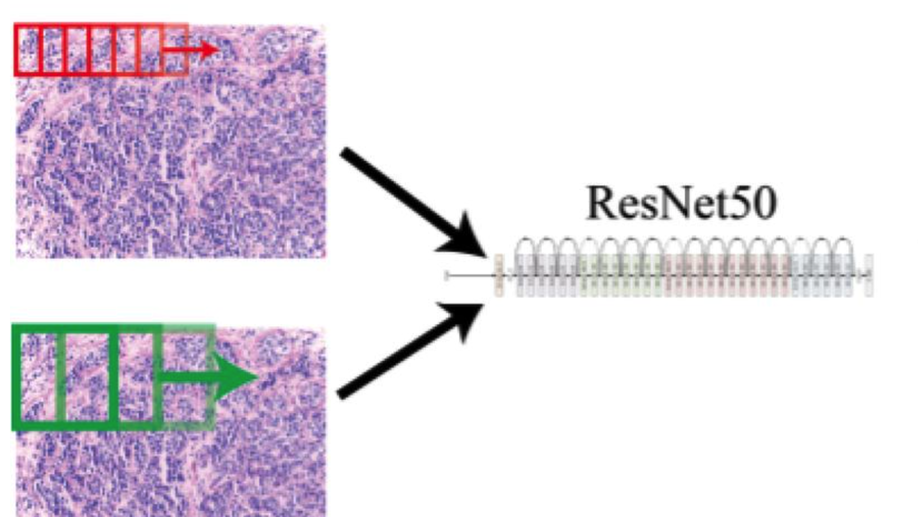
Convolving Pre-Trained Convolutional Neural Networks at Various Magnifications to Extract Diagnostic Features for Digital Pathology
Deep learning is an area of artificial intelligence that has received much attention in the past few years due to both an increase in computational power with the increased use of graphics processing units (GPU’s) for computational analyses and the performance of these class of algorithms on visual recognition tasks. They have found utility in applications ranging from image search to facial recognition for security and social media purposes. Their continued success has propelled their use across many new domains including the medical field, in areas of radiology and pathology in particular, as these fields are thought to be driven by visual recognition tasks. In this paper, we present an application of deep learning, termed ‘transfer learning’, using ResNet50, a pre-trained convolutional neural network (CNN) to act as a ‘feature-detector’ at various magnifications to identify low and high level features in digital pathology images of various breast lesions for the purpose of classifying them correctly into the labels of normal, benign, in-situ, or invasive carcinoma as provided in the ICIAR 2018 Breast Cancer Histology Challenge (BACH).
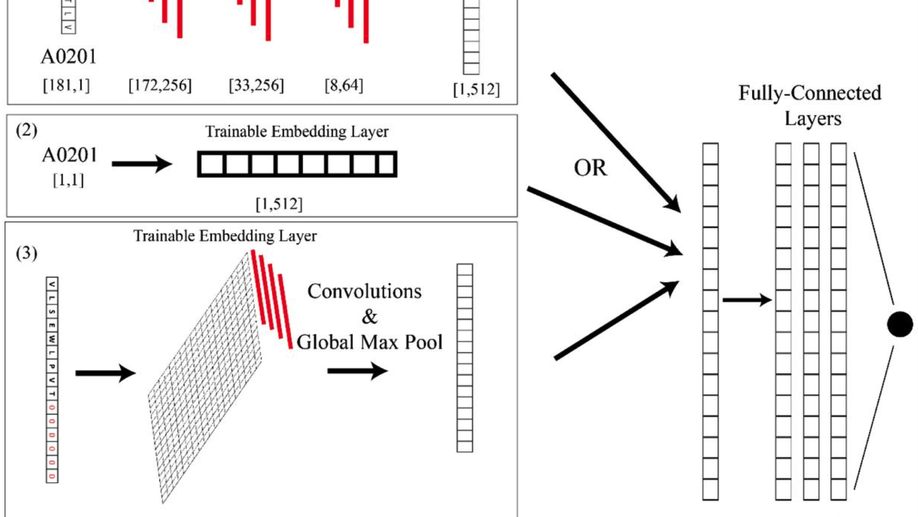
AI-MHC: an allele-integrated deep learning framework for improving Class I & Class II HLA-binding predictions
The immune system has potential to present a wide variety of peptides to itself as a means of surveillance for pathogenic invaders. This means of surveillances allows the immune system to detect peptides derives from bacterial, viral, and even oncologic sources. However, given the breadth of the epitope repertoire, in order to study immune responses to these epitopes, investigators have relied on textitin-silico prediction algorithms to help narrow down the list of candidate epitopes, and current methods still have much in the way of improvement.textless/ptextgreatertextlessh3textgreaterResultstextless/h3textgreater textlessptextgreaterWe present Allele-Integrated MHC (AI-MHC), a deep learning architecture with improved performance over the current state-of-the-art algorithms in human Class I and Class II MHC binding prediction. Our architecture utilizes a convolutional neural network that improves prediction accuracy by 1) allowing one neural network to be trained on all peptides for all alleles of a given class of MHC molecules by making the allele an input to the net and 2) introducing a global max pooling operation with an optimized kernel size that allows the architecture to achieve translational invariance in MHC-peptide binding analysis, making it suitable for sequence analytics where a frame of interest needs to be learned in a longer, variable length sequence. We assess AI-MHC against internal independent test sets and compare against all algorithms in the IEDB automated server benchmarks, demonstrating our algorithm achieves state-of-the-art for both Class I and Class II prediction.textless/ptextgreatertextlessh3textgreaterAvailability and Implementationtextless/h3textgreater textlessptextgreaterAI-MHC can be used via web interface at baras.pathology.jhu.edu/AI-MHC
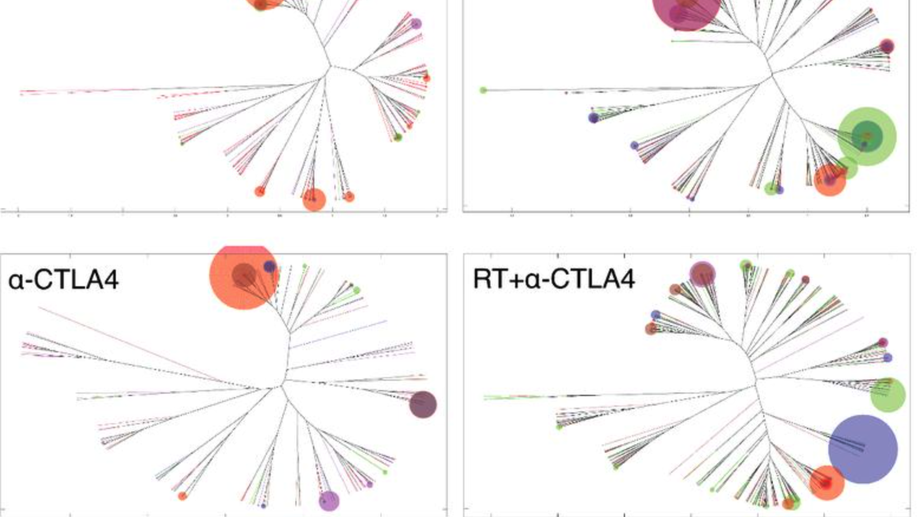
Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells
Immune checkpoint inhibitors activate T cells to reject tumors. Unique tumor mutations are key T-cell targets, but a comprehensive understanding of the nature of a successful antitumor T-cell response is lacking. To investigate the T-cell receptor (TCR) repertoire associated with treatment success versus failure, we used a well-characterized mouse carcinoma that is rejected by CD8 T cells in mice treated with radiotherapy (RT) and anti–CTLA-4 in combination, but not as monotherapy, and comprehensively analyzed tumor-infiltrating lymphocytes (TILs) by high-throughput sequencing of the TCRΒ CDR3 region. The combined treatment increased TIL density and CD8/CD4 ratio. Assessment of the frequency of T-cell clones indicated that anti–CTLA-4 resulted in fewer clones and a more oligoclonal repertoire compared with untreated tumors. In contrast, RT increased the CD8/CD4 ratio and broadened the TCR repertoire, and when used in combination with anti–CTLA-4, these selected T-cell clones proliferated. Hierarchical clustering of CDR3 sequences showed a treatment-specific clustering of TCRs that were shared by different mice. Abundant clonotypes were commonly shared between animals and yet treatment-specific. Analysis of amino-acid sequence similarities revealed a significant increase in the number and richness of dominant CDR3 motifs in tumors treated with RT + anti–CTLA-4 compared with control. The repertoire of TCRs reactive with a single tumor antigen recognized by CD8+ T cells was heterogeneous but highly clonal, irrespective of treatment. Overall, data support a model whereby a diverse TCR repertoire is required to achieve tumor rejection and may underlie the synergy between RT and CTLA-4 blockade. Cancer Immunol Res; 6(2); 139–50. ©2017 AACR.

ImmunoMap: A Bioinformatics Tool for T-cell Repertoire Analysis
Despite a dramatic increase in T-cell receptor (TCR) sequencing, few approaches biologically parse the data in a fashion that both helps yield new information about immune responses and may guide immunotherapeutic interventions. To address this issue, we developed a method, ImmunoMap, that utilizes a sequence analysis approach inspired by phylogenetics to examine TCR repertoire relatedness. ImmunoMap analysis of the CD8 T-cell response to self-antigen (Kb-TRP2) or to a model foreign antigen (Kb-SIY) in naïve and tumor-bearing B6 mice showed differences in the T-cell repertoire of self- versus foreign antigen-specific responses, potentially reflecting immune pressure by the tumor, and also detected lymphoid organ–specific differences in TCR repertoires. When ImmunoMap was used to analyze clinical trial data of tumor-infiltrating lymphocytes from patients being treated with anti–PD-1, ImmunoMap, but not standard TCR sequence analyses, revealed a clinically predicative signature in pre- and posttherapy samples. Cancer Immunol Res; 6(2); 151–62. ©2017 AACR.
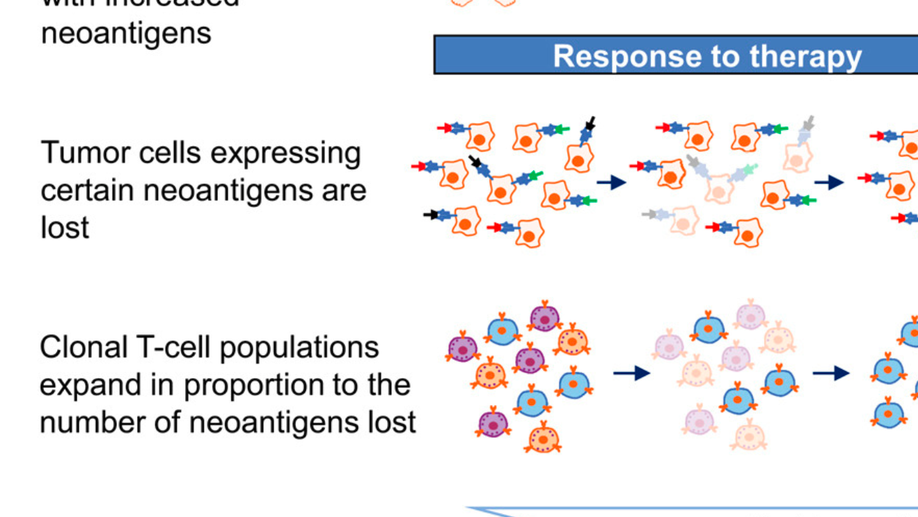
Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab
The mechanisms by which immune checkpoint blockade modulates tumor evolution during therapy are unclear. We assessed genomic changes in tumors from 68 patients with advanced melanoma, who progressed on ipilimumab or were ipilimumab-naive, before and after nivolumab initiation (CA209-038 study). Tumors were analyzed by whole-exome, transcriptome, and/or T cell receptor (TCR) sequencing. In responding patients, mutation and neoantigen load were reduced from baseline, and analysis of intratumoral heterogeneity during therapy demonstrated differential clonal evolution within tumors and putative selection against neoantigenic mutations on-therapy. Transcriptome analyses before and during nivolumab therapy revealed increases in distinct immune cell subsets, activation of specific transcriptional networks, and upregulation of immune checkpoint genes that were more pronounced in patients with response. Temporal changes in intratumoral TCR repertoire revealed expansion of T cell clones in the setting of neoantigen loss. Comprehensive genomic profiling data in this study provide insight into nivolumab’s mechanism of action.
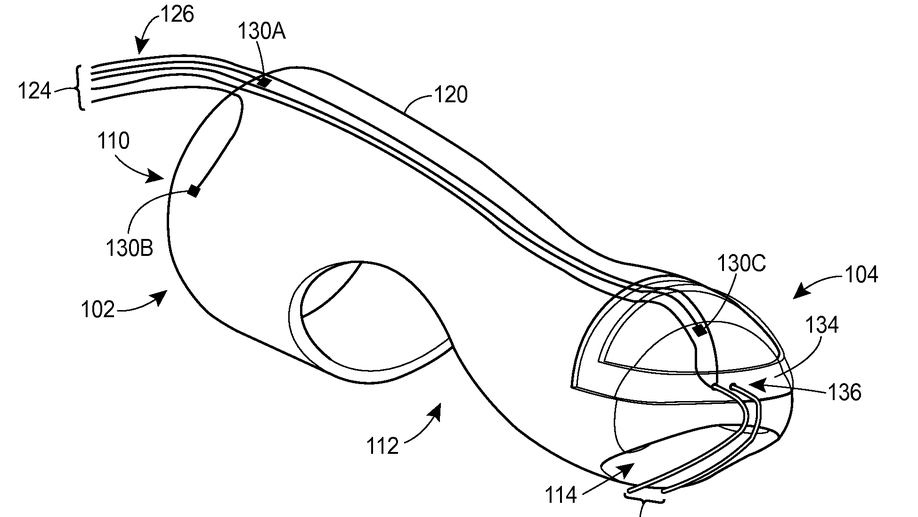
Digital manometry finger-mountable sensor device
A probe system includes a finger-mountable housing having a distal end and a proximal receptacle end. The proximal receptacle end defines an opening to receive a finger. The probe system also includes a probe assembly disposed on or within the finger-mountable housing and having at least a first sensor. The first sensor is positioned to measure a physical characteristic of a first tissue when the finger-mountable housing and probe assembly are inserted in a rectum of the patient.

Cryotherapy device and method for the treatment of cervical precancerous lesions
A device for providing a cryotherapy ablation treatment includes a piping assembly and a snow horn adapted to create a spray of snow from a pressurized source of a low-temperature liquid, a tubular applicator for collecting a mass of snow at a prescribed density that is sufficient to allow the mass to serve as the needed, low temperature, thermal reservoir for the device after the applicator’s distal end has been disconnected from the snow horn end so that it can to be used during the treatment process, and an applicator tip adapted to allow it to connect to the applicator’s distal end and be used to treat those specific locations which are to receive this treatment.
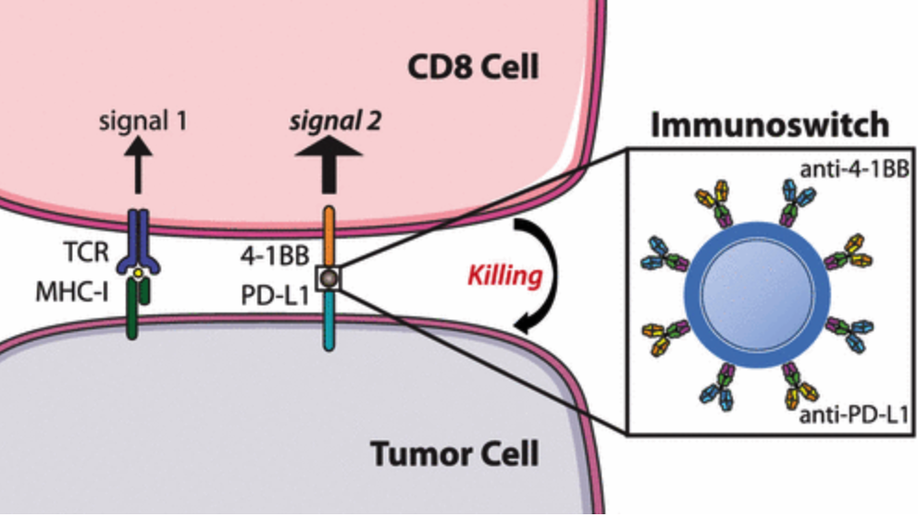
Dual Targeting Nanoparticle Stimulates the Immune System To Inhibit Tumor Growth
We describe the development of a nanoparticle platform that overcomes the immunosuppressive tumor microenvironment. These nanoparticles are coated with two different antibodies that simultaneously block the inhibitory checkpoint PD-L1 signal and stimulate T cells via the 4-1BB co-stimulatory pathway. These “immunoswitch” particles significantly delay tumor growth and extend survival in multiple in vivo models of murine melanoma and colon cancer in comparison to the use of soluble antibodies or nanoparticles separately conjugated with the inhibitory and stimulating antibodies. Immunoswitch particles enhance effector-target cell conjugation and bypass the requirement for a priori knowledge of tumor antigens. The use of the immunoswitch nanoparticles resulted in an increased density, specificity, and in vivo functionality of tumor-infiltrating CD8+ T cells. Changes in the T cell receptor repertoire against a single tumor antigen indicate immunoswitch particles expand an effective set of T cell clones. Our data show the potential of a signal-switching approach to cancer immunotherapy that simultaneously targets two stages of the cancer immunity cycle resulting in robust antitumor activity.
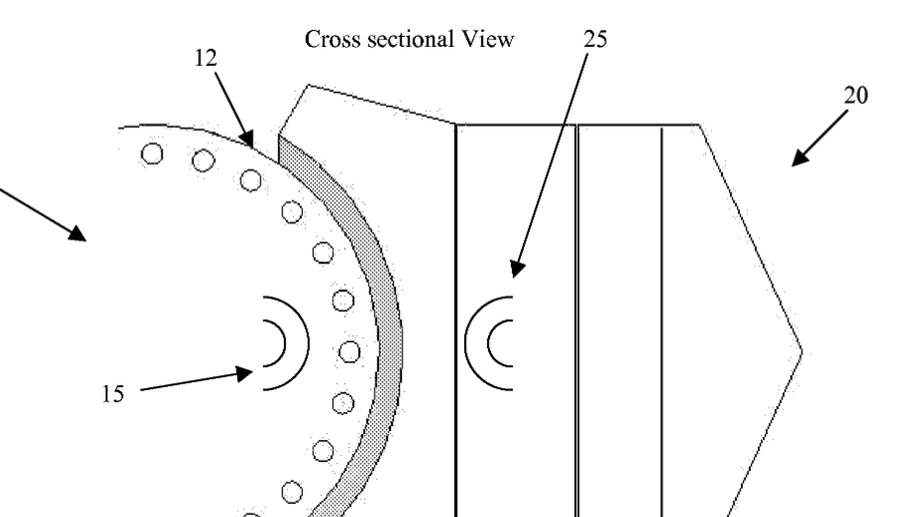
Device and method for force management within a joint
Disclosed is a device and method of management of forces within a joint. The device includes a first component with a first magnet arrangement providing a first magnetic field, a second component to interface with the first component with a second magnet arrangement providing a second magnetic field, and a compressible volume that is coupled with the second component that controls the separation of the first and second magnetic fields based upon a compressive force that causes the compressible volume to compress. The method includes using the normal force generated between the first and second components during joint use as the compressive force, causing the compressible volume to compress, bringing the first and a second magnetic fields into contact and overlap, and creating forces to couple with the normal force and regulating the overall normal force between the first and second components.