Welcome!
Thanks for stopping by and checking out my personal page. I am a clinical fellow in hematology & oncology at Weill Cornell Medicine, NewYork-Presbyterian Hospital. Prior to joining Weill Cornell for fellowship, I completed an MD/PhD from the Johns Hopkins University within the School of Medicine and Whiting School of Engineering, followed by my internal medicine residency at the Mount Sinai Hospital in the Icahn School of Medicine.
Doctoral Work
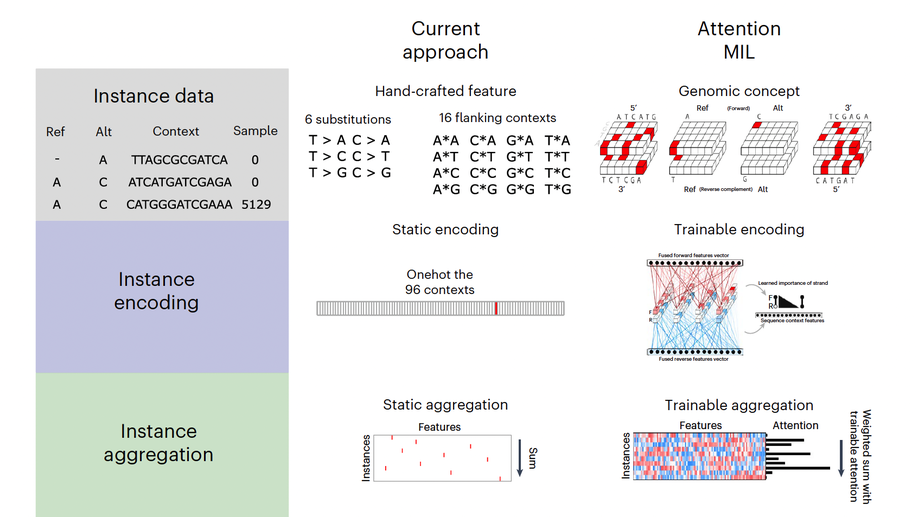
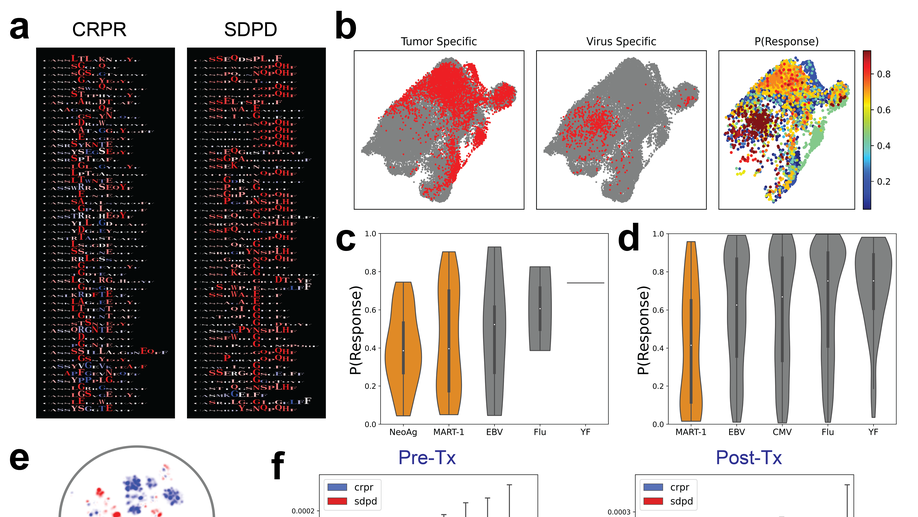
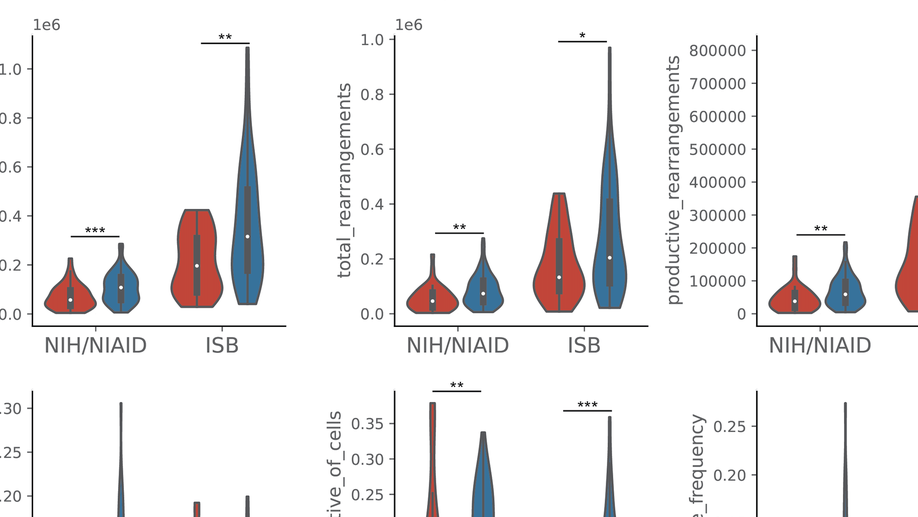
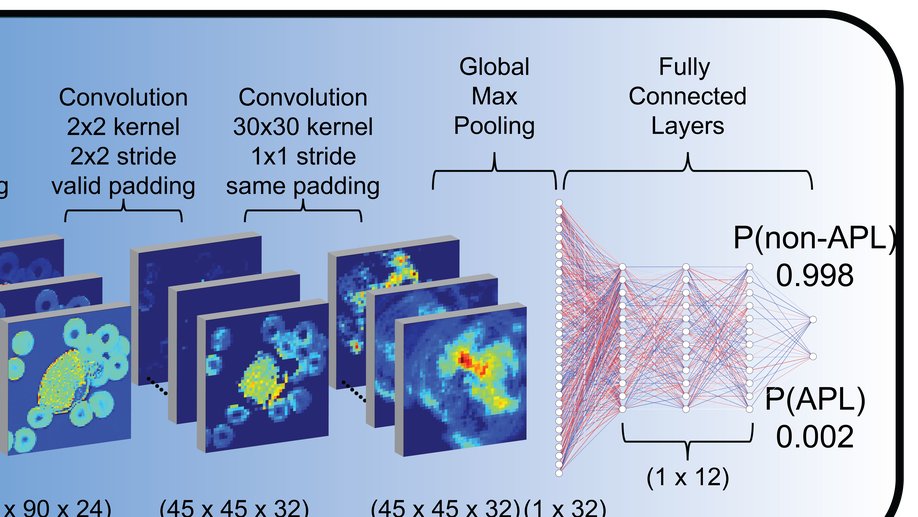
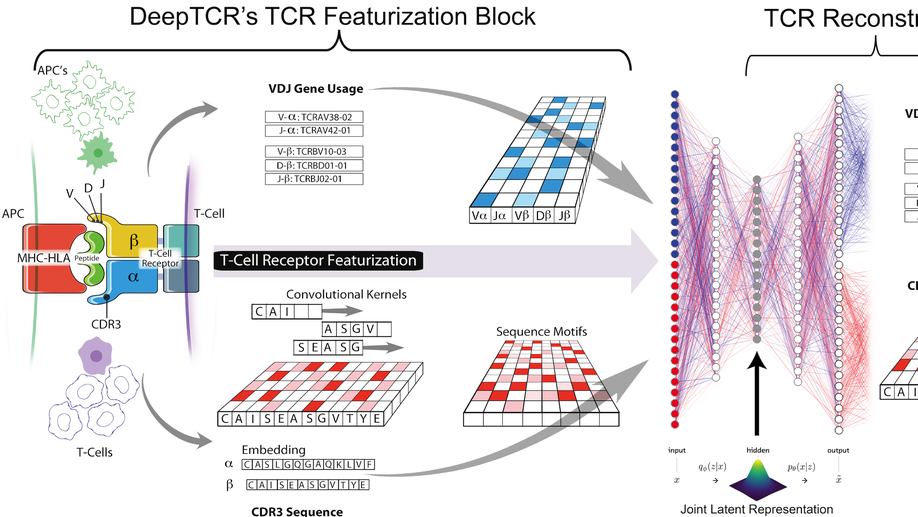
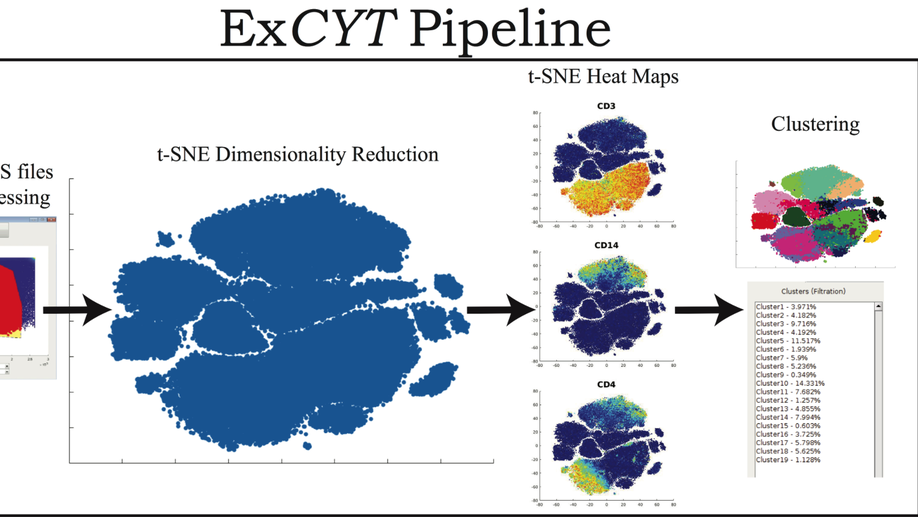
My doctoral work (Thesis Advisor: Drew M. Pardoll, MD.,PhD., Co-Mentor: Alexander S. Baras, MD,.PhD.) was at the intersection of artificial intelligence, machine learning, and cancer immunogenomics where I worked on several projects applying and/or developing novel algorithmic approaches to yield insights into the interaction of cancer and the immune system.
Pre-Doctoral & Master’s Work
Prior to matriculating into the MD/PhD program at Johns Hopkins, I completed a Bachelor of Science in Engineering (BSE) in Biomedical Engineering with a minor in mathematics and certificate of entrepreneurship at the University of Michigan in 2011. Following my undergraduate degree, I went on to complete a Master’s of Science & Engineering in Biomedical Engineering within the Center for Bioengineering Innovation and Design at Johns Hopkins. During this time (read about my experience in CBID here), I developed an interest and focus in medical device design, applying methods in computational mechanics to invent devices such as the CryoPop, a low-cost cryotherapy unit that utilizes readily available carbon dioxide tanks for the treatment of cervical cancer. The CryoPop has been issued a patent from the USPTO, completed its first clinical trial (NCT02367625), is currently enrolling in its second one (NCT04154644) to demonstrate efficacy, and is commercially available for clinical use through Pregna International Ltd.
Medical Mission
I also serve on the board of the student chapter of the Coptic Medical Association of North America (CMANA) as its current treasurer. As part of the student chapter, I have helped organized our annual conferences and local events that allow medical trainees at all stages to experience medical missions through the mother organization. Our student chapter also organizes opportunities for networking including online talks/forums, an annual gala, and quarterly newsletter.
Hobbies & Interests
When I am not coding away or in the hospital, I enjoy staying physically active (CrossFit, Olympic weightlifting, skiing, and most recently, triathlon), playing and producing music (singing, piano, guitar), and amateur photography. Otherwise, I enjoy visiting new cities, eating good food, and having a fun time with friends, family, & my dog Leo.
Contact Me!
I hope this page provides insight into the work I have done and areas of research I hope to continue to pursue in my future career as well as my own personal interests! Feel free to contact me and reach out with any questions or research collaboration opportunities!
Interests
- Cancer Immunology
- Artificial Intelligence
- Deep Learning
- Immunogenomics
- Internal Medicine
- Medical Oncology
Education
Doctorate of Medicine, 2021
Johns Hopkins University School of Medicine
Doctorate of Philosophy in Biomedical Engineering, 2021
Johns Hopkins University Whiting School of Engineering
Master's of Science & Engineering in Biomedical Engineering, 2012
Johns Hopkins University Whiting School of Engineering
Bachelor of Science in Engineering in Biomedical Engineering, 2011
University of Michigan
Deep Learning Nanodegree
Udacity School of AI